
Family Stories
Glimpses of the Glut1 Deficiency experience.
We hope you find something new yet also something familiar when you take a peek inside some of the special stories from our community.

Thank you to all who have participated and shared your unique and inspiring journey in the world of Glut1 Deficiency. These stories provide solace to families and enlightenment to the medical community. Your help plays an important role in our efforts to raise awareness and spread education.
We hope these stories bring smiles to your faces and add warmth to your hearts, for they are indeed glimpses of exceptional families and amazing children.
We want to hear from you! Share YOUR story with our community! You never know who needs to hear it. We feature stories on our social media pages along with our website. Fill out our online form or send inquiries to [email protected].
stories from our heroes:

Allison
Allison’s journey began at one month old. Just little things missing that had Mom concerned. “There’s something different about this baby.” With Allie being our fourth child, you just know. At three months we saw her first seizure. We didn’t recognize it at the time, and the doctor found nothing wrong. Nine weeks later, she had her first grand mal. We videotaped it and the search for a diagnosis began.
She was eight months old when we first heard the words, “Glucose Transport Defect.” We started our part in research with Dr. De Vivo and would travel to NYC whenever asked. We’ve maintained her health through the ketogenic diet. Though not a perfect solution, she prospered and made great strides. Walking was finally conquered at four years old. She has speech therapy at school as well as some outside therapy.
All her milestones are delayed, and at 15, she is just beginning to comprehend her disorder and has gotten over her embarrassment of it and now does her best to answer questions herself. She curses the diet but realizes the consequences when not followed. Her seizures last approximately 45 minutes and the postictal phase last five hours. That’s the toughest part of all for us as it’s hard to watch her suffer through it with little we can do but to make her as comfortable as possible.
Allie is always praying for a cure so she can eat French fries and pizza like the other kids and wants to drive, fly, be a doctor and a mommy! She wants to travel, work, and have a husband, a family and we want these things for her. She’s a great kid, mellow, caring, funny and on the honor roll….Go Allie!
2018 update
Allison is 23 now and with this aging has come a few changes. She has graduated from high school and attends a day program now where she is among many peers that she went through school with. She is picked up and dropped off at our front door daily. They take her out in the community where she has many different experiences from simple shopping to working at local businesses for an hour or so at a time. Recently she worked at a coffee shop doing custodial work and also at a Good Will type charity where she puts clothes on hangers and price tags. They teach math skills, kitchen skills, and do field trips. On Fridays they have a talent show and lately, Allie has been the dj, when not doing a Michael Jackson performance of her own. I’ve been told she pumps up the crowd and turns everything into a party!
In her down time, she participates in special Olympics bowling, and basketball, and belongs to DuMyon Martial Arts, where she is a blue belt in Tae Kwon Do. This is a non-profit organization that caters to those with IDD, etc. We originally joined TKD for self defense as clients in the day program can lash out for many reasons and she seemed to be in the wrong place at the wrong time, so she needed to learn how to deal with the unpredictability of others. She has been practicing for the 2020 Paralympics in Japan for over year. Last month we learned that TKD was being dropped from the Paralympics due to lack of interest around the world, and she proceeded to seize for 3 of the next 5 days as a side effect from this dream being dashed. Since joining TKD, she’s been released from Physical Therapy. She topped out at speech therapy, with them feeling they’ve done all they could. Her speech is hard to understand and she needs to be prompted to slow down and annunciate, which she will gladly do.
Her seizure activity includes 20-30 absence seizures a day. They do not disrupt her. We used to deal with seizures brought on by sneaking food, but locked cabinets and cameras helped her break that behavior. Most seizures now can be contributed to over excitement or huge disappointments. I still withhold carbohydrates whenever something big is happening in her life to avoid seizing.
On 8/3/12 Allison had a grand mal while swimming and I couldn’t get her out of the pool. During the rescue her head was dropped hard on the cement and we lost her. She was gone for three minutes and was Flight for Life’d by helicopter from Colorado Springs to Denver Children’s Hospital where she was put into a medical coma for three days. She was back in that same pool three weeks later having no memory of the ordeal and was completely recovered in six weeks. On the 5th anniversary, she got a tattoo on her wrist to commemorate the experience and to remind herself that life is precious.
Other interests include dancing, swimming, hiking, traveling, haunted houses and horror flicks! She is thinking of going to college for acting and sign language. She has had some modeling opportunities, which she loves. Allison is very adventurous and game for anything. She is in a long term relationship and hopes to marry some day. He is autistic and together, they need to work on independence but there is no way of knowing whether together, they can manage on their own. They would love to drive and have their own place someday, but she is aware that she has to be seizure free to get a license, and has so much to learn to take care of herself and a home. We always speak of the future in an opportunistic, positive way.
We have had to find new doctors as she is now 23, and its been easier as the years pass to do so. After puberty her diet was changed from the Ketogenic to Modified Atkins as petit mals were coming too frequently. As a child, her seizures were mostly grand mals lasting 45 minutes, but now are usually petit mal that last for a minute or two and come in small clusters. She no longer loses consciousness, or is incontinent during these episodes, and vomiting is rare now. She feels better faster with two Tylenol, O2, and a good nap. Her new neurologist has heard a heart murmur and she has an appointment in April to have that checked out.
Allison is loved by everyone. She has an easy going personality, a great sense of humor, and I’m incredibly proud of her and her strength and resilience! She is eager to please and too trusting of humanity, making her vulnerable in public and on-line. But she’s never alone as she’s medically fragile. Her daytime para’s always hope to have her in their group as they say she’s “so easy!”

Addie
Addie was born in December 2011. To all, she appeared to be a normal, healthy little girl. For a number of reasons, Addie came to live with Deb and I – her grandparents. As she grew, we noticed that she missed some of the normal developmental milestones. She was late talking, and late crawling, but there is a range of normal so we were not overly concerned.
In October, 2012 we were driving across north Dallas when my wife looked back at Addie and noticed some peculiar behavior. Her head was moving back and forth, and her eyes were also jerking. We have since learned that these are typical early symptoms of Glut1. My wife was afraid she was having a seizure so we rushed to a local hospital that had a children’s unit. By the time we got Addie into the ER, the jerking had stopped and she appeared to be normal. The hospital kept her overnight with an EEG hooked up and monitored. After 24 hours, there was no abnormal brain activity so the doctors discounted the possibility of a seizure. She was discharged with no diagnosis.
Addie started walking at 13 months in January 2013. Within weeks of walking, she started falling and was unable to get up. There was no warning or alerting behaviors prior to these episodes. One evening, Addie started having an episode and my wife recorded it on her phone. We spoke with her Pediatrician and showed her the video, but she had no idea. This happened again, so we took her to Dallas Children’s ER. By chance, the Director of the Epilepsy Unit was there and saw the video. She thought it was Episodic Ataxia. We told her that the episodes appear to be triggered by heat or physical exertion. Addie spent two days in the hospital hooked up to an EEG. They tried to provoke a seizure using lights and found nothing.
At some point, Addie had a DNA test. The results were inconclusive, yet did show some abnormalities in a couple of genes, one of which was associated with ataxia. Neither my wife or I had ever heard of this condition, so I did a little research and it did look like Addie’s condition. She was placed on the normal drug for that condition, but it did not stop the frequency or the duration of the episodes. After 6 months, I contacted the National Ataxia Foundation to find a specialist . They referred me to Dr. Abigail Collins at Denver Children’s Hospital.
We made an appointment to see her and, in October 2013, we met with doctor Collins. We shared this history with her and the results of the DNA test. She talked to Addie, took her for a long walk and then told us that, in her opinion, Addie did not have Episodic Ataxia but had Glut1 Transporter Deficiency Syndrome. She said the only way to be 100% sure was to have a lumbar puncture. We asked about the syndrome and were provided with a little information about a condition we had never heard of, and some information about the Glut1 Deficiency Foundation.
We returned to Dallas and made arrangements for Addie to have the lumbar study done. The following January, the test was run. While Addie was still in the procedure room, her Neurologist came and told us that it was definitely Glut1. Her blood glucose was normal, but her spinal glucose was at 33 – about 30% of normal.
Based on these findings, the Ketogenic Diet was prescribed. We had a chance to meet with and discuss Addie with Dr. Pascual, who examined Addie and reviewed her history. He agreed with the diagnosis and the treatment with the diet, but he told us that the diet is pretty harsh on the body and must be started during a 3 day stay in the hospital. In March 2014, Addie started the diet. On her second day, she fell into what appeared to be a coma or extremely deep sleep immediately after a meal. She could not be woken. The nurses worked and tracked her vitals and eventually she did come back. We’re still not sure what happened, but obviously her young body was having a negative reaction to ketosis. This prompted an extra day as an inpatient. But, by the end of day 4, she was normal and in ketosis.
She has now been on the diet for 3 1/2 years and has had no recurrence of the ataxia. She goes in for blood work 3 times a year and seems to be progressing normally. Last year, my wife Deb passed away from brain cancer. We all miss her, but especially Addie. She tells me everyday that she misses her Nana. It was Deb who kept pushing the doctors for an answer and one day Addie will know how much her Nana loved her.
This year Addie wanted to meet other Glut1 kids. The York family in Oklahoma held a fundraiser in October. Since that was only a few hundred miles away, we went and Addie got to meet Abby. She loved that short trip. Then a few weeks later, a Glut1 child from Pennsylvania was due to meet with Dr. Pascual in Dallas. Addie got to meet and play with Millie at a local McDonalds.
I’m so grateful to Erin, Millie’s mom, for making this meeting possible. Since Glut1 is so rare, these kids sometimes feel like they are the only ones. It’s wonderful that Addie had the opportunity to meet two other little girls who face her same challenge.
Addie is now in Kindergarten and may be showing some signs of cognitive delay. She has always learned very quickly from what she hears. But, I have noticed a problem with visual letter and number identification. Since she is just now beginning to read and write, these issues weren’t evident before. Her Neurologist thinks this apparent lack of ability is probably an effect of Glut1.
We just had our annual recheck with Dr. Pascual. I told him that her movement symptoms are still under control with the diet, but she was showing some signs of cognitive delay or deficiency. She can’t seem to learn how to recognize letters or numbers. She has no problem with word meaning or the concept of numbers, just can’t recognize them visually. He said that he had hoped that she could avoid these cognitive components of Glut1, but sadly these are common observations with our kids. Hopefully, this is merely the sign of a delay and not something more complicated.
Addie will undergo some psychological tests this spring to determine if they are indeed Glut1 effects or if she possibly just has abnormal delays in some cognitive processing abilities. The tests may also indicate if there are other deficiencies yet to manifest themselves, or if these issues will be overcome in time.
Even with this latest concern, I am very thankful that I found doctors who know what Glut1 is, how to identify it, and how to treat it. I am also grateful that Deb and I found the Glut1 family who give us all support, information, and helpful history. This will give Addie the best chance to live a normal, healthy, and productive life.

Ayla
Ayla is 13 years old and in seventh grade. She was diagnosed with Glut1 almost two years ago. We didn’t really notice anything abnormal until she was about four years old. She would occasionally have dizzy spells or so we thought they were dizzy spells, she was also very uncoordinated and unbalanced at times. For us we just thought maybe she was clumsy .
When she started kindergarten is when we really started noticing the delays in school compared to the other kids. At first the school tried to tell us that they thought she had ADHD because of her not really paying attention much, and when there was a lot going on in the classroom was when we started to notice the dizzy spells happening more often. Every time we would take her to the doctor or the hospital for the dizzy spells by the time we got there they would be done and over with and they were just saying maybe it’s all inner ear related .
As her kindergarten year went on we decided that it would probably be best for the fine motor skills to start occupational therapy, that helped with handwriting but needed physical therapy for the balance and coordination. By age seven she still wasn’t reading which we knew maybe it was a vision problem, come to find out her eyes would not focus at the same time together so we then started vision therapy at Good Shepard hospital. Although it was masking most of the problem it was also helping her learn how to read because it was focusing her eyes. We finished an 18 month course of vision therapy and she was able to read from left to right with no problem and the words were not bouncing all over the page. We took a break from the program and then A little while later Covid hit, School was shut down and she was on an iPad all day and that is one when we really started noticing the spacing out and more dizzy spells.
Between our doctor for the vision therapy and then seeing a physiatrist it took a very long time to have someone actually give Ayla an EEG. My husband and I researched and realized that we thought she was having seizures. It was really starting to affect her every day activities. When her EEG results came back her doctor told us that we needed to see the Children’s Hospital. They also did an EEG and then an overnight EEG which came back that Ayla had 64 absence seizures In 24 hours. They immediately put her on Ethosuximide twice a day and then tested her through genetics and found that she has a mutated gene and has Glut1 .
My husband and I were relieved to have finally have some answers to what was going on and she fits every symptom and although her case is not as severe as others this has let us down a path that has changed our lives. With finally having a diagnosis we were able to get Ayla help with everything that she needed and a neuropsych evaluation for learning disabilities. She is now enrolled in a small private school and is doing fantastic. The seizure medication has made her seizure free for two years, and with modifying her diet a little bit she feels great.

Carson
Our son Carson started developing abnormal eye movements around 3 months old. We were taken via ambulance when these eye movements started during a pediatrician’s office visit. His doctor thought he was having a focal seizure. Unfortunately, this was just the beginning of a long, exhausting journey. Our hospital stay showed no seizures on his EEG and a normal MRI, so we were dismissed and told that he was “normal.” I wasn’t sure how I was supposed to live knowing our baby was NOT normal, but the doctors said he was, so I wearily accepted it.
From there, his eye movements continued periodically. I would try to ignore it, but I knew something was wrong. He was a happy baby otherwise, who met all of his milestones on time.
A few months later, he had a grand mal seizure, and it seemed like this was the kickstart to unleashing so many more symptoms than we ever thought possible. He started falling, a lot. His legs would randomly buckle and his arms and torso would uncontrollably flail out. By the time we got to the closest ER, the episode would be over. It was incredibly frustrating and exhausting. His body would appear to be lifeless at times, he would have absent seizures, he would experience half paralysis, and tons of movement issues that we had never seen in our lives. He had issues with fine motor coordination, speech and cognitive delays, and pretty severe lack of impulse control.
During this time we worked closely with his neurologist. He had genetic testing done, tons of EEGs, brain MRIs, CT scans, urine analysis, lots of labs, and spinal taps. To our horror, one of his CT scans actually showed a tumor in his lung. We began treatment with oncology and thought we had our answer.
We were, however, wrong.
Almost two years of autoimmune therapy, the disease they suspected he had was not responding normally to the standard treatment. We couldn’t believe it, but after all that time we had found ourselves yet again on the path of an unknown diagnosis and tons of questions.
After it became apparent that his autoimmune treatment wasn’t working, we decided to switch gears. His neurologist ordered another spinal tap to check on the previously low glucose levels. It was once again, abnormally low. This time it was compared to the blood serum level at the same time and the difference was indicative of Glut1 Transporter Deficiency Syndrome. We also worked with a lab in France to test Carson’s blood. By the grace of God, his samples were safely sent overseas and they were able to distinctly see that his blood did not properly transport glucose. We finally had a for sure diagnosis.
The ketogenic diet became an answer to our prayers. At times it has been a battle and heartbreaking, but knowing the difference between the child who was not on the diet and the child who now is – words cannot describe. His energy level is so much better, he has endurance, he isn’t seizing and he isn’t falling. We used to have to leave so many meet ups and play dates because of Carson’s legs not working. That isn’t happening anymore! While we continue to pray for a complete cure for this disease one day, we are still counting our blessings. Every day we have with Carson is a gift. He is learning so quickly, excelling in language arts, and is an incredibly social 6 year old. It has been a LONG and exhausting road, but he has persevered through it all. He always reminds us, in his words, to “never, ever give up.” He is that motto in living form.

Chris
Christopher was born on December 22, 1997 after a somewhat rocky pregnancy. His mom had placenta previa. His labor was rapid after being induced. His apgar scores were 9 and 10. We first started noticing something was not right when he was about 5 months old. It was really hot outside and we were at Lowe’s. He got limp as a dish rag and was “out of it. “ This happened a few times. He also had paralysis episodes. At 9 months, he was not able to sit and was not hitting his milestones. His eyes were crossing and we knew that something was wrong. He was diagnosed with Cerebral Palsy and the journey began. He was in First Steps and did therapy 5 to 6 times per week. He really started to make progress. At 18 months he was walking but shortly after that, we noticed these odd little head “drops.” He would have them in the mornings mainly but it became so frequent that he was having at least 100 a day of these little drops. This was the beginning of a long journey to a diagnosis.
After trying every possible seizure med which made him worsen, we begged to put him on the ketogenic diet. We were told that it was only for severely mentally handicapped children and that he would need a G tube. We weren’t willing to accept that so we took him to Johns Hopkins where he started the keto diet by mouth at age 3. He was so toxic on medications that he got severely dehydrated and lost so much weight that we had to take him off the diet. We knew that it was working for his seizures but he was very sick. After 6 months and a weight gain, the seizures got worse again so we took him to Chicago and had him placed on the ketogenic diet. It worked-IMMEDIATELY. He was talking, running and so happy. It was our miracle. This went on for 2 years and we tried to take him off. The seizures came back. So we tried again for another 2 years and again, took him off the diet and the seizures returned. It was a vicious cycle and we had a lot of physicians, family and friends doubting us and thinking that we were crazy for keeping him on the diet for so long. But in our guts, we knew it was the only thing that worked, we just didn’t know why.
After being extremely depressed and frustrated, we called Jim Abrahm’s with the Charlie Foundation (at 6 am-oops) and he suggested that we contacted Beth Zupec Kania in Wisconsin. He gave us her contact information and we contacted her. A few days later at about 9 pm, she called back and we told her our story. She asked if Chris had ever had a spinal tap and we said, no. She suggested that before we take him off the diet and consider brain surgery, we might consider having him tested for the Glut 1. This was the first time we had heard it. We called our neurologist and they did not take us seriously. So we had our pediatrician call another neurologist and he ordered the tap. A week later and at age 9, Chris was diagnosed with Glut 1. If it weren’t for Beth, I don’t think we would have been diagnosed. But who knows, right? We came to meet Dr. DeVivo and his team a few months later and fine tuned Chris’ diet.
Chris is now 12 years old. He is known for his million dollar smile. He loves Purdue and collects baseball hats. He can ride a bike for miles at a time. He is a boy scout. He loves to Play Wii, trade silly bands, look at cars and has a love for life. He has 3 brothers who keep him in check. He has 6 grandparents, 16 cousins and 4 aunts, 5 uncles who all love him very much. If his dream were to come true, he would be able to eat like everyone else. Chris has taught many in his short life that we can’t take life for granted. We are so blessed and should live each day to the fullest. We are so fortunate to have him in our family.
update 2011
Chris completed 6th grade this year and will go to middle school next year. He continues to surprise us all with his reading and math skill development. His biggest milestone this past year was his social development. Chris has a best friend now! His name is Chris, too! He has 2 good friends from school and they have had play dates and overnights. This has really helped Chris blossom. Another exciting thing for Chris this year was getting a puppy. We got “Henry” for the boys on Easter weekend. It is truly beyond amazing to see the love that our sweet lab puppy can share with Chris. Chris feeds Henry and is the first to take him out every morning. Henry is so patient and loving. We could not have asked for a better “companion” for him.
Chris continues to be on the ketogenic diet. His seizure control has not been as great this year but mostly due to Chris becoming so very tired of the oil and cream. He has been on the diet for 10 years now. We are very proud of him for being such a trooper!
update 2018
Chris is now 20 years old. He graduated from high school with a certificate of completion in 2017. He is a busy man that works at Chic-Fil-A, Good Will and volunteers at various community activities. He attends a social day program group twice a week to hang with his peers. He is involved in Special Olympics where has participated in swimming, basketball and getting ready to start cycling. He is involved in Best Buddies and also at his church. He loves taking the city bus where he needs to go and enjoys Ubering as well.
Getting to 20 wasn’t easy for Chris and puberty was difficult to say the least. He developed a movement disorder which is common with Glut1 and got ketoacidosis which slowed him down for over two years. Chris has an awesome team (dietician, MSW, doctor and case manager) that developed a plan for him to learn to remain on the ketogenic diet as an adult using an exchange system with menu cards and it has worked very well for him. He also has used an app on his phone to track his energy and keeps fat bombs with him as needed to maintain energy. With a combination of the diet, supplements and medication, he is doing much better now and is thriving.
Chris recently went to the “Night to Shine” and had a blast. His family is continuing to work with Chris to develop a “Good Life” as an adult life for Chris with many different connections, challenges and opportunities. Glut1 is not going to slow Chris down. We are very thankful to the Glut1 Deficiency Foundation for its continued work in advocating and creating awareness in the Glut1 community.

Cleo
Cleo was my long awaited second child, born in November of 1995, via a normal, easy birth delivered on her due date. As a loving family we were so happy to bring Cleo home. Her 8-year-old big brother was excited to have a sister. He took on his big brother duties well. Cleo was a restless baby who was breastfed frequently on demand, as she never settled for very long. At the age of 8 weeks old, we noticed what were Cleo’s very first symptoms of Glut1 Deficiency Syndrome which included vacant looks (absence seizures), odd arm twitches (myoclonic jerks) and abnormal eye movements (aberrant gaze saccades). This was an uncertain time for us as a family. As a mother I recognized Cleo’s symptoms were unusual, but naively, I wasn’t overly concerned. I had assumed medication would solve the problem if required.
After first consulting our local GP with these episodes, we were referred to a Pediatrician when Cleo was 7 months of age. Cleo’s general development at this time seemed on target. She was sitting, crawling, and cruising at 9 months and able to walk unaided at 15 months.
Cleo’s Pediatrician seemed to draw a blank when I explained these unusual episodes. This was until he witnessed an abnormal eye movement at a clinic appointment himself. From there, Cleo was referred to a Neurologist at the University Hospital Wales in July 1997. Cleo was now noted to have developed a complex movement disorder. These episodes were longer and easier to videotape for medical reference. She appeared wobbly and fell over frequently, became floppy, but also had bouts of spasticity where her limbs were stiff. She had problems with controlling all movement when these episodes occurred. Seizures presented as absences, myoclonic jerks, tonic clonic seizures, and speech arrest, none of which responded to AED’s. Episodic symptoms seemed to be getting increasingly worse affecting her daily. Head drops had become routine on waking. Ataxia, spasticity, hypotonia, hemiplegia and tremor became worse when triggered by exercise, hunger, illness or excitement. She was generally very lethargic, continually tired and falling asleep, and her concentration was very limited. The earlier naivety I had once felt had, by this time, been replaced by stark fear of the unknown. Cleo’s symptoms were unusual to us and frightening. At this point our medical team were running tests to eliminate possible life limiting conditions.
At Cleo’s routine 2.5 yrs. assessment, speech and language skills were reported to be below the referral line for therapy. Manipulative and self-care skills were also below, so a referral for Physiotherapy, Occupational, and Speech therapy was to ensue. We were drawing blanks for a confirmed diagnosis and we needed a second opinion. At this time, a referral was made to Great Ormond Street Hospital London, where they concurred with Cleo’s local consultant that Cleo had a likely Ion Channel Disorder. It was a vague diagnosis with no further treatment options. This explanation of Cleo’s symptoms and events leaves out the worry and heartbreak that goes with living with an undiagnosed incorrect medical condition. The drug trials that don’t help, the hospital admissions, EEG’s, MRI scans, endless tests leave an indelible mark on the family. We carried it well, but as I write this some 20 years later, the feelings haven’t left. I can still locate the fear I felt in my stomach to this day. “What was wrong with my little girl?” We lived each day alongside the unpredictability of symptoms and we tried to manage the special occasions that always seemed blighted by extra symptom breakthroughs.
Then we had a change of fortune. Cleo’s Neurologist retired and her new Neurologist, Dr. Sandeep Jayawant, wanted Cleo to be tested for a rare medical condition called Glut1 Deficiency Syndrome. Eventually, a lumbar puncture was performed showing hypoglycorrhachia. The samples were sent for Glucose uptake assay to Prof. Dr Jörg Klepper who practiced in Germany. This led to a confirmed diagnosis of Glut1 Deficiency Syndrome on December 13, 2002, when Cleo was 7 years old. We had finally turned a corner with diagnosis. I cannot understate the relief we felt as a family to finally have a path that provided direction when we had felt vulnerable and adrift for so long. With diagnosis, the knowledge, services, and support opened up, along with one of the most comforting aspects which is the connection to other families who share the diagnosis. From the point of diagnosis, we were advised the treatment was the Ketogenic Diet which they said would be difficult, or we could try to supplement Cleo’s regular diet with Corn Starch, in theory to push carbohydrates and increase energy. Our hospital had little knowledge of the diet at that time and even less of Glut1 Deficiency Syndrome. Our first attempt with the Ketogenic Diet transition was difficult. We were admitted to the hospital for a 3-day stay. The food choices were extremely limited and Cleo’s symptoms became uncontrollable in the transition phase between reducing glucose and not producing ketones. Our medical team advised us to stop the initiation of the Ketogenic Diet due to Cleo’s severe symptom reaction, and to instead supplement Cleo’s normal diet with Corn Starch which proved to have very little help with symptoms.
We wanted support with the new diagnosis and found it in the charity formerly named C.L.I.M.B, now Metabolic Support UK. At that time, there were no other UK families that had been diagnosed with Glut1 DS. They searched the internet and made contact with Dr. De Vivo and Kristin Engelstad at The Neurological Institute of New York Columbia University Irving Medical Centre, New York, who invited us in April 2005 to take part in a Glut1 DS research project, whereby they identified Cleo’s missense mutation T1371, not identified in either parents. It was a huge privilege to meet with Dr. De Vivo MD and his team. They helped us understand the diagnosis of Glut1 DS and, with their encouragement, we left feeling that we must try to initiate the Ketogenic diet again, this time being more prepared through the knowledge we had gained. Our other real milestone of the trip was meeting our first-ever family who shared Cleo’s diagnosis.
It was an absolute pleasure to meet Matt Rizzo and Janet Bean, with whom we continue our friendship to this day.
On returning from the USA, we obtained another referral to start the Ketogenic Diet once more. Two weeks short of Cleo’s 8th Birthday, we initiated the Ketogenic Diet at home this time with full medical dietician support. The transition was very smooth in comparison to the first trial. Cleo produced ketones quickly and the results were miraculous, and to this day it astounds me how a Ketogenic Diet can have such a positive impact on symptoms. I would encourage anyone with doubts to get support from your medical team and Ketogenic services to give your child the chance of improved quality of life. Cleo continues to use the Ketogenic Diet as the best form of treatment to control her symptoms. We are forever grateful for its positive effects. Puberty was a particularly difficult time and, unfortunately, the diet has never been as effective since. We have explored alternative options to help regulate Cleo’s hormones and monthly cycle including various contraceptive pills and the contraceptive implant. We have found the Merina coil to be of some help. New cycles of symptoms continue to come and go, strange itching sensations in hands, a numb tongue, abnormal facial movements, PED’s (Paroxysmal Exercise-Induced Dyskinesia), Athetosis (involuntary twisting and writhing movements) Dysarthria, (unclear speech articulation) Dystonia, (involuntary muscle contractions that lead to abnormal posture and movements). Pain and cyclic vomiting persist to this day. These are the particularly difficult symptoms to witness as a family. Cleo’s determination to handle these episodes are inspirational.
Matthew’s Friends Ketogenic Dietary Therapies have been very supportive of our UK Glut1DS community, providing dietary support while combining professional consultations and social events together. In August 2013 at one of these events, we combined a private clinic appointment at Matthew’s Friends Clinics with Dr Klepper. We discussed the difficulties Cleo was having with symptom control, ketone production and puberty. He suggested as a family we gain knowledge around the possibility of Cleo having a gastrointestinal tube to aid ketone production and independence. We went home and thought long and hard about the decision, and we concluded that it needed to be one Cleo would ultimately make. This was a difficult decision for Cleo as a young woman, however we believed we had tried every option possible to tweak Cleo’s oral diet and gain back stability. Ultimately her strength of character and “get on with it” attitude made her decide she had nothing to lose. At 22-years-old in February 2018, Cleo had the procedure for a PEG (percutaneous endoscopic gastrostomy). Since the procedure, we have gained good ketone production and Cleo is able to manage her dietary requirements independently when well.
Unfortunately, Cleo continues to struggle with absence seizures, nausea, and vomiting which is an ongoing problem for her and her medical team. We have formed a wonderful relationship with Cleo’s Neurologist at UHW, Dr. Johann te Water Naude, and the Dietitian team at Bristol Southmead Hospital, who form the foundation of her treatment and care.
Cleo’s education from Nursery to College was met in mainstream with a full time 1-to-1 Learning Support Assistant. Education brings with it another set of difficulties, but overall Cleo enjoyed the company of the other students and she made some particularly strong relationships with her LSA’s over the years who provided support and kindness. Cleo is a social young woman who loves being with people. She’s strong and direct, She also enjoys time alone watching her favorite tv programs and catching up on social media.
As her mum, I’m astounded by her resilience. I’m so proud of the woman she’s become. She never gives up! This is merely an account of dates, symptoms, and events. Cleo is far more than this description. As a family, we have made wonderful connections with our global Glut1 DS community, particularly through the support we have found with the Glut1 Deficiency Foundation. We have attended both the European Glut1 Deficiency conferences in Milan and London, several events for Glut1 DS families held by Matthews Friends in the UK, and most recently the Glut1 Deficiency Foundation conference in Washington D.C., whereby she represented Wales at the red-carpet event. We are fortunate to have medical experts working on behalf of our Glut1 DS community. We hope with new breakthroughs in research, alternative treatments will be found. We regularly join the G1DF Zoom meetings where Cleo has found support and fun amongst her peers and I have found friendship and connection through volunteering with the G1DF.
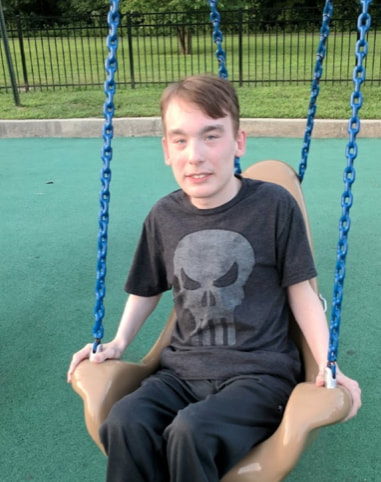
Colin
Colin was born in January 1999. He was a preemie weighing only 3 lbs 14oz. Despite being a preemie, he was in good health and needed very little assistance from the NICU. Colin was allowed to go home after only 10 days in the hospital. When Colin was about 3-4 months old, he began having seizures. In the beginning, he was experiencing a seizure once every few days. The doctor put Colin on Phenobarbital and the seizures progressively got worse. By the time he started the diet, he was having up to 150 “episodes” a day.
Our road to a diagnosis began in April 1999 and the roller coaster ended in October of that year, when Colin was diagnosed with Glut-1 DS, an extremely rare condition. At the time of his diagnosis, there were only 11 known cases in the world. The doctors told us that there is no cure to fix this condition but, there was a treatment. The treatment is the Ketogenic Diet. The hope is to be on the diet and have good seizure control without the aid of medication. The diet is also to help his brain grow and to not be deprived of energy. Colin started the diet on October 29, 1999 at St. Christopher’s Hospital in Philadelphia. We saw immediate results. From 150 seizures to Zero!
Colin came home from the hospital, He was doing very well. They took him off the medicine and we focused on the Ketogenic diet. Early interventions stepped in to help with physical, cognitive and speech therapies in the home until Colin started school on Jan 25, 2003. Colin was reaching milestones of sitting, crawling, walking and babbling. He was a very happy boy.
During the winter of 2004, when he was just 5, Colin had malrotation of his intestines and needed major surgery to correct this issue. The recovery was long. Colin was given a G-Tube placement in his abdomen. He started to be able to eat solids 3 weeks after the surgery. He was not happy about it. He fought eating all of the time. Due to the complexity of the diet, the Dr. suggested we use the Keto formula. This would help while he was trying to regain the ability to eat again. Colin is still currently on the formula and tube fed. He never regained the ability to eat foods by mouth.
In 2010, Colin was doing well but having a difficult time with everything. He could not communicate any of his needs. This was frustrating for him and he would act out when he was not happy. The doctors started to do lots of testing to see if they could pinpoint what was wrong and why he was upset. Sometimes he would cry for hours. After 8-10 months of testing, Colin was diagnosed with ICP. This is intracranial pressure that acts as if a tumor is in the brain. (Never documented or diagnosed in a child with Glut1) A Spinal tap was done to relieve the pressure and the results were immediate. My happy boy was back again.
In 2012, due to his fragile bones, Colin broke his ankle while getting off the bus. This recovery was very long and immobilized Colin for some time. This was also the time he was going through puberty. During his recovery, he was in a wheelchair for almost a year. This set him back a lot. I believe with each medical issue we regressed as if we had never made any progress. He stopped doing many things he was doing in the past and began to just sit and not do anything. This was a very hard time for him.
In 2013, Colin was diagnosed with Scoliosis. His spine and hips were turning and it made it difficult for him to walk. We pushed forward and continued to do physical therapy to get him mobile again. By 2014, he was walking only short distances and was still struggling as his body was much bigger.
In 2015, Colin was screaming and in pain a lot. He would hold his head and cover his eyes. Instead of doing a full work up of tests, I asked the Dr. to just do the spinal tap to check the pressure of the brain fluid since ICP is reoccurring. Colin had ICP again. Once the pressure was released, he was back to normal. We are praying ICP does not return. If it does he will need a shunt inserted to prevent future ICP occurrences.
By 2016, Colin was also diagnosed with Kyphosis which is a hunch in his back. He struggled walking and standing upright. The doctors informed us that his bone deformities would continue until he was done growing. Still, he continued to be a happy boy and worked hard to be able to walk without the aid of a wheelchair or walker.
In 2018, Colin was healthy and happy most of the time. His family had a huge birthday bash for his 18th birthday and he loved every minute. He started a new school and received in-home behavioral therapy which helped him make so many strides. He currently is still functioning on a 2 year old level. He needs assistance in bathing, toileting, feeding, dressing, and all basic self-help skills. He rarely sleeps normal hours. We find Melatonin, a sound machine, and a weighted blanket work well for him to sleep, but he still has nights of no sleep and headaches. He is non-verbal and still struggles to grasp a form of communication. He can answer some questions with picture exchange, but still has no real communication and would not be able to communicate in the outside world.
He has balance issues, walks dragging his foot, and cannot stand upright. But, he is still walking! Colin is able to follow one-step directions. He loves to watch his siblings play outside and go on the swings. He loves movies, music, and anything that lights up or spins!
We went through guardianship for Colin and have made many preparations for his life as an adult. Our family has made many advances in the care of Colin and we will continue to do whatever we can to ensure he will have a life full of love, happiness, and good health. We hope one day to see the cure for this disorder, but we do know Colin’s case is on the more severe side of the spectrum. He has always maintained good seizure control and for this we are thankful.

Colton
This is the life story of our now 18 year old son on his journey to find answers to a condition that caused our family incredible fear and stress as he was growing up.
When our son Colton was an infant he began rapid eye blinking. A hospital visit determined that this was our sons quirk for going to sleep. When Colton was around 2 years old, my sister mentioned how often he seemed to fall down, something I had chalked up to his age. When he turned 3, our son Colton began falling down. His legs seemed to crumple beneath him. He was unable stand or pull himself up. After a rest period (normally as we raced to the emergency room), he would regain his normal function. Due to this quick turn around, doctors were baffled by what was plaguing him. We were even scoffed at when asked by a emergency room doctor if he was just a clumsy kid.
By the time Colton was 4 years old, we were being followed by a paediatrician, neurologist, and a family doctor. We were told many things over the next 14 years. Some of the diagnoses he was given included water in his ear, stomach tumour, muscle dystrophy, vertigo, and childhood migraines. It was terrifying each time a new symptom would emerge.
As he got older his cognitive development began showing signs of impairment and the gap between him and his peers was becoming larger. He was dwarfed compared to this classmates standing 2-3 feet shorter than most of them. His fine motor skills were so weak he required tables to type on due to his inability to write. Even his voice seemed forced. His pupils stayed fixed at an unusually large size. His gross motor skills continued to decline. A wheel chair was purchased to have on hand for him. As his teen years hit, he was suffering from daily migraines which turned into hemiplegia migraines (simulating stroke symptoms). After two rounds of genetic testing, results continued to be returned as normal. At this time we had already sought a second opinion from a new neurologist but still didn’t feel like our son was diagnosed correctly. We applied to be seen by Toronto’s sick kids hospital, but were turned down. We then made the difficult decision to cancel our family trip to Disney and applied to the Mayo Clinic. We were thrilled when we were accepted fearing this was our last chance. After two days of driving and a 2 hour consultation, we were told they too were unable to help us. We were devastated.
Finally, with special permission we had a third round of genetic testing done. This time it was finally revealed that our son was living with a rare form of Glut1 Deficiency. Although it was suggested to reduce his carbs to 20 a day, it was quickly determined that this did not help his symptoms.
Today Colton is 18, turning 19 years of age at the end of April. He has graduated high school and lives with his mom, dad, brother and sister. We have had to fight to get him reassessed to qualify for ODSP and did. He is on a waitlist for Community Living and assisted living (waitlist is 10y ears). He is a happy man who loves movies, trivia and bowling. He continues to have daily headaches, but has fewer instances of falling down (unless he’s been waking for extended periods of time).
We spent all of our son’s childhood wondering if/when we would lose our son. It was the most terrifying and painful time in our lives. We hope by spreading awareness that our son can help prevent other parents from going through the horrors we experienced.

Dante
Dante is 5 years old. He has a twin brother named Dominic. Dante was born at 4 lbs 6 ounces, and his brother at 6 lbs 6 ounces. Dante had marginal cord insertion while he was in the womb, so he did not get all of the nutrients that he could have. We had to have multiple ultra sounds to make sure he was growing. Other than that, it was a normal pregnancy.
We did not notice much of a difference in development until Dominic started to roll over. It took Dante quite a bit longer, but he was determined to do it! When Dominic was able to sit up, crawl, and walk, we noticed that Dante was also delayed. He eventually caught up. He was a strong and determined baby! When the twins were about 1.5, we noticed him very off balance. I always described him as a drunk penguin. He was also not speaking clearly at all. It was very easy to spot these delays because his twin brother was always right on target. We brought Dante in to be evaluated after his physician suggested it. He was evaluated for autism. They said he did not have autism, but he could benefit from Speech therapy, Occupational therapy, and Physical therapy. We got him enrolled in all of the therapies. He is still enrolled in all three, and loves it all. The therapists all say he is always happy and willing to learn.
When he was about 3 years old, he started having seizures. It looked like he would go out of consciousness for a quick second, and then he would be back to his normal self. He would be walking, and then his eyes would roll back and his head would slowly go down. Sometimes he would fall if it happened while he was walking. It was very scary for all of us. We got an MRI and an EEG. Nothing came of it. All of the doctors that have been evaluating Dante had finally diagnosed him with DCD (Developmental Coordination Disorder) and Epilepsy. For DCD, there is no cure. They told us to continue the therapies. For the seizures, he was prescribed Lamotrigine. Since the meds were not working, they adjusted the dosage. Still no change, but we continued to have regular check ups with the Neurologist. We talked to biogenetics, and they did some blood tests. Nothing came out of that. After multiple tests and a lot of unanswered questions, we were asked if we wanted to do some Genetic testing. We happily agreed to that.
The genetic test came back, and we finally received an actual diagnosis. He does not have DCD. We found out he has Glut 1 Deficiency Syndrome. This diagnosis explains all of the concerns we have about Dante. We were so happy to FINALLY get some answers, and a little frustrated that it took so long. Nonetheless, we are happy to start the journey of healing and we are very hopeful for Dante’s future.

Dara
Hello, I’m Ailbhe and along with Adam my husband and Sam (5) and Dara (3) we are the O’Reilly family from Dublin, Ireland. Dara has Glut1 deficiency syndrome and was diagnosed at 19 months old.
He had his first obvious symptom at around 7 weeks old when he had 2 non-breathing episodes. He had a lumbar puncture at this time but Glut1 was not considered. More symptoms appeared including paroxysmal eye movements and he was diagnosed with myoclonic epilepsy of infancy at 6 months old. Due to his developmental delay, hypotonia and epilepsy he had a genetic test at 15 months old and that’s how we got our answer. It was bittersweet but we were so happy to find answers and such a supportive community. He started on the classical keto diet shortly after and we noticed he was much more alert and took a great interest in books while in hospital. He instantly became seizure free.
And we all lived happily ever after…..
Well happily enough-last April after 17 months of seizure freedom he is having seizures again, worse than before. Dara now walks independently, has had a speech explosion and uses some 3 word sentences and he understands similar to a neurotypical child of his age. He is very sociable and fun loving. He looks up to his big brother Sam who doesn’t fully understand his condition yet.
We had such a great sense of achievement that we were able to attend the Glut1 conference in San Diego with Dara who absolutely loved the traveling, surprisingly, as he is a creature of habit! We look forward to what the future holds for Dara and his GLUT1 friends.

Dominic
Dominic was born in September 2010. When Dominic was 5 months old, he began having short episodes of rapid eye movements with a one-sided gaze. This happened twice in a month’s time and we saw a Neurologist at Cooper University Hospital in Camden, NJ. He was not medicated at the time. His Neurologist ordered an MRI and a 5-day ambulatory EEG. Both were normal. On May 5, 2011, Dominic had a grand mal seizure. This was his first and only one to date. Dominic was started on Keppra. In the meantime, Dominic seemed to be reaching appropriate milestones.
The rapid eye movements progressed to rapid eye movements coupled with weakness of the whole body and lip smacking. It always would seem to happen at mealtime in his high chair. We would have periods of time where there would be no “seizure” activity and then we would have a hard few months. At 12 months of age, we started worrying about Dominic not babbling and not pulling himself up to walk. He also never seemed to be able to entertain himself. He couldn’t even sit in a bouncy seat for a few minutes by himself to play. The Pediatrician was not concerned about development nor was his Neurologist. Dominic walked at 18 months of age, but not well. He was unbalanced and had many falls. He also was not speaking, only making non-verbal gestures and “huh” sounds for things he wanted. I began seeking physical therapy and speech therapy. Although we probably needed occupational therapy as well, his fine motor deficits never seemed to be as remarkable.
Dominic’s seizure activity then progressed to left-sided weakness episodes (as if he had had a stroke) and episodes in which he would not be able to move his body at all. He might lean to one side and not be able to hold himself up, his gait would worsen, and he would get pale and agitated. Again, I always noticed this was worse at meal times and he would be better after eating a meal or drinking milk. Lamictal was then added along with the Keppra. My biggest concern began to be how Dominic’s milestones would regress after a seizure. He would finally learn to do something, have a seizure, and not be able to do it again for a period of time. Dominic’s Neurologist was concerned about this as well and decided to do a metabolic lab work-up. We had another negative MRI and negative 3 day EEG. Dominic had abnormal metabolic lab studies but nothing that would correlate with any specific condition. It was suggested to us that we see a Developmental Pediatrician for an evaluation as well. This evaluation showed the delays we already knew about and nothing more. We began feeling like Dominic was just going to be who he was. We needed to stop looking. But I never felt completely comfortable with having abnormal metabolic labs and not getting another opinion. So we saw Metabolic/Genetic specialists at Children’s Hospital of Philadelphia, had genetic lab testing performed and Dominic was diagnosed with GLUT 1 in July 2013. An LP supported the diagnosis.
This news was both devastating and a relief at the same time. We started the ketogenic diet in September of 2013 and he has made miraculous progress. Some improvements came immediately and others took some time. His attention span was the first thing to improve. Initially, his posture was more relaxed and he didn’t look “hazed ” anymore. He was finally able to try to do the things a 3 year old should be able to do. Now Dominic’s gait is completely normal when walking and slightly ataxic when running. He does have a tendency to fall, but much less than he used to. Self-feeding is always a struggle but not because he can not do it, more so he doesn’t want to. Dominic is now full of life and energy. His speech and verbal abilities have exploded. He speaks like a typical 7 year old. Normal vocabulary with some articulation delays. Dominic built a castle out of blocks 7 days after being on the diet. He had never done this before. Now he builds LEGO sets! Dominic is in first grade and reading on a first grade level. His math skills are at first grade level as well. He is in a learning disabled classroom because he learns differently or sometimes a little slower than other kids. He struggles with writing and fatigues quickly. He has good days and bad days, but mostly good days. Sometimes if he is sick, or sometimes for unknown reasons, his left leg will appear weak. His gait will be altered and he will seem like he is having a hard time performing all functions. Usually hydration and feeding him will help and then the symptoms will pass within a day or two. The difference for us on the keto diet is that after Dominic has an “episode,” his milestones do not regress as they did prior to the keto diet. I would say our biggest struggle with Dominic is that he is easily fatigued by exercise; especially walking long distances. Heat seems to make this even worse. We do not let this stop us. We find ways for Dominic to participate in everything. We choose indoor activities when it is hot out and we use a stroller/wheelchair for when he can’t walk a long distance.
Dominic is the bravest boy we know. We can’t imagine what he has gone through in his early years of life and how unbelievably happy and loving he has been. He rarely gets frustrated when trying to communicate, he loves being with other children, and when he can’t do something because of his disabilities he either lets it go or tries to do it again.
Looking back and now understanding his diagnosis, there were clues since birth. I nursed him for 5 months and Dominic over fed. He was hungry every 30 minutes to 1 hour (even at night). I stopped nursing because of this and thought that bottle-feeding would help. This is when he began having seizures. Over time, Dominic was a grazer in the cabinets and looking for food all the time. His body was requiring glucose more often than normal to help get more glucose to the brain. Our Neurologist now thinks that most of Dominic’s “seizures” must have been hypoglycemic episodes of the brain.
Dominic is an incredible little boy who seems to accept anything, fights for everything, and is truly the focus of our lives and the drive to get us out of bed in the morning. He loves his family. He especially loves his sister Iris, his dog Ruby, and his trains! His sister (who is now 14) has been unbelievably nurturing to Dominic. She lives and breathes for him and wants nothing more than his love. He has changed our lives in countless ways. We have embraced the challenges we were given in life and want to make nothing but the best of our journey. Our family focuses on our blessings and positivity. It may be challenging and sorrowful at times, but we are hopeful Dominic will continue to flourish and touch the hearts of everyone he meets.
Update May 2022:
Dominic is now 11 years old. He has been on the ketogenic diet for 8 years. We have transitioned from a 3:1 ratio to a 2:25:1 ratio. We tried a 2:1 ratio and Dominic ended up having difficulty concentrating, behavioral issues, and unsteady/floppy gait. The diet has become easier to handle over the years as we have figured out ways to integrate some keto products on the market into Dom’s meals.
Dominic no longer has ataxia at baseline when walking or running. He will occasionally have a bad day and seem to fall a lot. Dominic seems to be sensitive to elevation and will have PED’s after flying. He gets a migraine occasionally and symptoms are headache, difficulty seeing or blurry vision, and fatigue. We have been able to manage this at home with Motrin and sleep. His ketones are never low when the migraine occurs.
Dominic is in a 5th grade mild to moderate learning disability class. He still receives PT and OT on a consult basis (frequent check-ins) through the school. He receives speech therapy mostly for his language delays, reading comprehension, sequencing, and expressive language skills. Dom has good articulation. Dominic gets writer’s fatigue and struggles with complex math. We have started utilizing the calculator and dictation programs. He has tested at a 2nd grade reading level and a 4th grade math level.
Dominic is very active. He plays golf, karate, video games, and has learned to ride a two-wheel bike without training wheels. Dominic has lots of friends at school and in our neighborhood. He is motivated to be independent. We are slowly working on independence with the ketogenic diet, but feel as if we have a long way to go.
Dominic is such a happy loving child. We are hopeful that he remains mostly asymptomatic and that he can continue to tolerate the keto diet as long as he can.

Drake
Drake was born in August of 2009. Our family’s Glut1 journey started shortly after Drake turned three years old. Early warning signs started at the age of two, but they were just little things that seemed off. Drake would get super silly and goofy and then his gait would become jerky and uncoordinated. He would talk slowly, especially when nervous or excited, with long pauses between words. He had trouble grabbing small objects and didn’t seem as dexterous as other two year olds. Sometimes he almost seemed tipsy.
As a baby, he hit all his milestones. He was crawling at 8 months and walking by 15 months old. His temperament was easy going and relaxed, and he was very alert and observant of his surroundings. He was born healthy and happy without any complications after an uneventful pregnancy and birth.
We started becoming concerned about his speech when he was 16 months old. But right after the age of two, he experienced a language explosion and began combining words to make simple sentences. This was paired with an intense curiosity about the world around him, especially in regards to how things worked. We called him our “little observer”.
He had three memorable episodes of exercise-induced dyskinesia (sudden involuntary leg spasms) at the age of two. It worried us, but he didn’t seem to be bothered or hurt by it. It made him super silly and goofy. We wondered if maybe he just laughed himself to the point he couldn’t walk or maybe he was moving his legs on his own?
His three-year-old wellness visit uncovered low-muscle tone, microcephaly (small-head circumference), balance issues (unable to stand on one foot for more than a few seconds) and trouble with jumping and landing on his feet. When I mentioned the leg spasms, we were advised to see a Neurologist right away. This popped our bubble that it was simply due to either growth spurts, a cold, or an ear infection. We were also encouraged to start OT and PT therapy.
The first Neurologist diagnosed Drake with gait-ataxia and ataxia. The journey then began to find the underlying cause. MRI and blood tests results all came back negative. The next possible step was a lumbar puncture to check his Cerebral Spinal Fluid (CSF), but unfortunately we balked at doing that. When we finally did it years later, it revealed moderately low CSF glucose levels and confirmed the diagnosis of Glut1 DS that we had just received a month prior via genetic testing.
Drake’s episodes of ataxia, dyskinesia and slurred speech increased from a couple of times a month to a couple of times a week between the ages of three to five years old. Episodes would last from ten minutes to over an hour and would often occur in clusters. He did OT, PT and Speech Therapy and Drake constantly improved and achieved the goals set for him, being at most between three to six months behind his peers. He was good-natured about his attacks of “wobbly legs” and “drunken sailor walking,” smiling even when he was unable to control his legs and lower body. Attacks would usually occur in the late afternoon or evenings when he was most tired. He would always recover after a snack and a chance to rest. From the time Drake was a baby, I found that he was happiest if he ate every two to three hours when awake, and even to this day I never leave the house without a snack for him.
Our second opinion Neurologist eventually diagnosed Drake incorrectly with Episodic Ataxia (the disorder), and after a while he began taking the medication Diamox three times a day. He took it for about a year total from the age of five to the age of six, but then stopped after starting the ketogenic diet — to his immense relief. The Diamox medicine, for the most part, stopped his movement episodes of dyskinesia and ataxia, but did nothing to address his energy levels, sensitivity to heat, balance, and coordination issues. We also noticed he was having trouble with double vision, dizziness, and nausea. But, we still don’t know if these were side effects of the Diamox or symptoms of Glut1 Deficiency that have now gone away with the ketogenic diet.
Thankfully we had genetic testing done when Drake was six years old. We did it simply to determine which type of Episodic Ataxia he had, EA1, EA2, etc. Thanks to the efforts of the Glut1 Deficiency Foundation, the Glut1 community, and doctors around the world, the SLC2A1 gene was included in his panel, and a mutation was found. And while Drake didn’t fit the more severe symptoms, we immediately knew this was the right diagnosis. It finally explained all of his symptoms: his movement disorders, his slow slurred speech, his fine and gross motor delays, his being easily fatigued and overheated.
Drake has now been on the ketogenic diet for over two years. He is eight years old as of 2/1/18. Since the age of six, and his very first week of Keto, he has been symptom free of ataxia and dyskinesia. He took right to the diet as if his body was craving high fat foods. Drake loves his keto food and the fact that so many adults around him are also doing keto.
He is in the regular second grade class at school and getting good grades. We are very lucky that he probably appears typical to most people, but of course he has to work harder than his peers in some areas, especially in regards to handwriting. His favorite subjects in school are math and science. He is reading at grade level and has no cognitive delays. He is a hard-working blue belt in Taekwondo and just started learning the Bo staff. He loves playing video games, especially Minecraft on the XBOX and Creativerse on the computer.

Eleanor
Eleanor is a spunky two-and-a-half year old. She was born at 32 weeks at 4 pounds 4 ounces and had a month-long NICU stay. At 6 weeks old, she began showing symptoms (spasms and two unresponsive episodes). She had her first lumbar puncture at this time with low glucose, shortly followed by two more LPs that confirmed those results. After genetic testing at 3 months old coming back negative for the SLC2A1 gene mutation, we were given good news that she didn’t have Glut1.
Eleanor began having more seizure-like episodes at 4 months old and was diagnosed with acid reflux instead by her Pediatrician. We later found out that all of those episodes were in fact seizures.
At 18 months old, Eleanor still wasn’t walking and began having rapid eye movements so, with Glut1DS still in the back of our heads, we requested further testing. After several MRIs, CTs, EEGs, and an additional LP and PET scan, it was determined that she has Glut1DS without the genetic component. She began walking at 19 months old.
In August 2021, Eleanor was put on a ketogenic diet. With the help of Speech and Occupational Therapy, her coordination has improved tremendously and she’s now talking more with 2-3 word phrases! She’s part of the Rare Genomes Project through the Broad Institute of MIT and Harvard, with the hope of helping other kids just like her with rare or genetically undiagnosed conditions.

Finnian
Our beautiful Finnian is our 5th child, our rainbow baby – born after we lost one of his big sisters, Nora in December 2019 to complications associated to her rare congenital defects – we were blessed with her for 6.5 months, we carry her in our hearts every day. Finnian’s other siblings are his brother Tiarnán, 7 and his big sister, Maeve, 5 and Nora’s twin sister, Clodagh now 3.5 year old.
Finnian was born at 37 weeks but monitored very closely for the last 9 weeks of the pregnancy because his growth slowed suddenty at 28 weeks, dropping from the 50 percentile to below the 4th for the rest of the pregnancy. He was a perfect but tiny 2.3 kg baby boy. Early on we noticed he seemed different to our other babies, but our history of trauma had us thinking we were being overly protective. He didn’t make proper eye contact, was slow to grow, and was slow with his early developmental milestones – he didn’t properly smile until he was 14- 15 weeks old, and these were rare when they happened. From about 10 weeks we started to notice strange behaviours with his eyes and then at 12 weeks he had his first seizure. After a hospital visit, we were told to monitor him and bring him back if we noticed anything more. Two weeks later he had his first extensive abnormal eye movement episode which lasted for over 10 minutes and rendered him exhausted. We rushed him back to the hospital thinking he had another seizure. Again we were sent home and told to monitor him.
Another eye episode happened a week later – this time I refused to leave the hospital until someone told me what was happening to my child. Thankfully he had a few more episodes in the hospital and I also had video on my phone – this time I couldn’t be brushed aside as an ‘overprotective mum who’d already lost a child’. We met with a wonderful neurologist who took us seriously and ordered a full set of tests including a Lumbar Puncture. The LP showed low sugar in Finnian’s spinal fluid which prompted Glut1 genetic testing. It would take a further month for the results to return and in that time our family caught Covid-19, which led to Finnian becoming very unwell and suffering with 24 hours of seizures. During this hospital stay, Finnian’s results came back positive for the Glut1 mutation. He was started on a Ketogenic diet within a couple of hours of his results coming back. Within a week of being in ketosis Finnian laughed for the first time, it was like we were watching our beautiful boy ‘wake up’ – almost like someone had turned the lights on. He has continued to thrive with the ketogenic diet and some support from a Physiotherapist and is now meeting all his age appropriate milestones, including just learning to walk now at 16 months. He has a little bit of dyskinesia with his tongue and fingers but nothing that is holding him back at this stage.
We are so grateful to have a diagnosis, and while we don’t know what his future will hold, we have every reason to believe he will live a full, happy, healthy life. The ketogenic diet does pose its challenges, particularly growing up with older siblings – (to be honest it can be downright heartbreaking sometimes), but we are learning every day how to manage and still allow Finnian to have a joyful relationship with food.

Gemma
Gemma had symptoms at a very early age, but Glut1 was not discovered then. We had nowhere to get help or support. She wasn’t diagnosed until she was 26 although she was on all sorts of seizure medications from a young age. She never complained. After diagnosis she was offered the keto diet which worked and it was amazing. Unfortunately we had to stop.
Gemma is amazing. She passed her exams and her driving test and had a baby. Her son is now 7 and does not have Glut1. We are so proud of her. She’s on seizure medication and takes life as it comes, but she still never complains.

Hailey
Hailey was born in April 2016. We were so happy to bring our princess home to meet her two brothers, Nathan and Elijah. For 7 weeks, all she did was eat and sleep. Because of her lethargy, we kept saying that something was wrong, but everybody, including the medical staff, said she was just a very good baby and appeared to be normal in every way.
Then, our lives changed forever. Hailey began to have seizures, but I didn’t yet comprehend what was going on. I decided to take her to the Pediatrician near where we lived because I thought she had aspirated some milk while I was feeding her. By the time we arrived at the doctor’s office, her heart rate was 240+ BPM, and the doctor agreed that we should immediately take her to the ER. While in the ER, tests were performed and then we were transported to Memorial Hermann Children’s Hospital at the Texas Medical Center south of downtown Houston. At this point, she was seizing nonstop. After about 80 seizures, they gave her some Ativan to stop them. The next morning, the medical team started Hailey on Keppra and did a spinal tap while we waited for answers.
After reviewing the test results, the doctor told us the diagnosis was GLUT 1. It was a lot to take in, and we were in denial because it is such a rare condition. How could she have this? Hailey was sent home after being on Keppra for just 5 days, and we were advised to come back in about 3 weeks to start the ketogenic diet. Two days later, we had to call 911 because I couldn’t get the seizures to stop. At this point, she was so weak that we couldn’t get her to eat anything. An NG feeding tube was inserted so we could provide her with the keto diet and medicines needed. We were in the hospital for 3 weeks, and she slowly began to improve.
We were still not convinced that her condition was GLUT1, so we underwent genetic testing and were told it would take about 6 months to get the answer we wanted so badly. But God was on our side, and we got answers in two months.
Yes it is GLUT1, and this is how our story begins.
Hailey is now 9 months old and is such a wonderful, happy little girl who brings joy to everyone who meets her. People look at her and say she looks good, but they don’t see the invisible condition that we are fighting on a daily basis. Yes, she looks to be doing well because she has God on her side, and He fights hard for her everyday. For such a little girl, she has gone through so much. She has had more blood drawn from her tiny body than I have in 32 years and has had more hospital stays than she deserves, BUT she is strong and so are we because of our faith in God.
She is on Keppra and Phenobarbital to help with the seizure control. She also is on the ketogenic diet, and we are still trying to find the right ratio that will work for her effectively. It seems that we are in the hospital about every 6 weeks for some kind of setback.
Hailey has a G-tube because it is so important that she takes in all her food and oils to keep her from seizing. She also gets all of her medicine, consisting of pills normally taken by mouth, through the tube . If it weren’t for her feeding tube, I don’t know how we would manage this situation. There are times when she doesn’t want to drink or eat, but unlike other kids she must have the nourishment and medications or she will become very sick. So yes, she looks good because we make certain that she doesn’t miss a single feeding time and also receives as much nourishment and medications as needed on a regular basis every day and at specific times throughout the day.
She gets PT, OT, and speech therapy every week to help her body to develop normally. We are blessed to have all of the therapists come to our house for Hailey’s sessions. They are a wonderful group of ladies that love her so much and they work hard to give her all the help she needs.
There is so much we don’t know about GLUT1, but thanks to the GLUT1 Deficiency Foundation, we have a support group to help us deal with this challenging medical condition on a daily basis. In Hailey’s case, GLUT1 is an invisible condition that most people can’t see, but it is so real to us. There is never a day without worry or concern that we are doing all we can do for her. We, as a family, wouldn’t have been able to cope with all of the unforeseen challenges involving Hailey’s medical situation without God in our lives. He has given us the peace that surpasses all understanding. He is with us all the time. He blessed us with early diagnosis so that she could get the right treatment right away, and we are so grateful for His love for our family.
We have a long road ahead of us but I know God will be there with us every step of the way. There is always hope for a cure.

Huxley
Almost two years ago to the day, I rushed to the ER with my three-month-old baby not breathing. After a few days stay and multiple tests (including the lumber puncture that showed low glucose but they didn’t connect that to Glut1 at the time) they discharged us saying “babies do weird things”.
Two weeks later it happened again. Huxley had 15 seizures in a row and no matter how many drugs they pumped into him, they didn’t stop. This time we were adamant we weren’t leaving without answers. Thanks to my husband’s research, he suggested genetic testing immediately to rule out Glut1. He had found a medical journal online after extensive reading and the symptoms matched our son. This time we left with a baby on seizure medicine that made him sleep all the time and his sweet smile was gone. We had what we thought was a 4-6 week wait for the results of the genetic testing.
A few days later Huxley had what we now know are “aberrant gaze saccades‘’. His little eyes were darting all over the room and it was terrifying. This time we went to a different hospital and this neurologist rushed the genetic testing. On New Years Eve 2020 at 5pm we got the answers we so desperately needed. Huxley has a missense in the SLC2A1 gene, he has Glucose Transport Disorder. On January 1st we started the medical ketogenic diet. Huxley went from breastfed to bottle fed with keto formula over night. Now, at 2 years old he is thriving. The diet was life changing for him.
The early diagnosis was everything. As a parent, I will always be advocating and raising awareness. That doctor could have diagnosed Huxley on the night of his first seizure.

Jacob
Jacob was born via c-section in March 2012. He was a very large but healthy baby. The second I held him I knew something was wrong, but I didn’t have any real reason to feel like that. At a few weeks old, Jacob was diagnosed with GERD and colic. By 6 months old he wasn’t hitting his developmental milestones. We began birth23 soon after that.
By the time Jacob turned one, he was a much happier child and doing well. That didn’t last long because a few weeks later Jacob had a seizure. From that point on things got much worse. Jacob had more seizure-type episodes, was becoming more behind developmentally, and had very low energy. Jacob would end up in the hospital a few times because of his “episodes” and still we had no answers. During all of this, Jacob was diagnosed with low tone and began wearing ankle braces. He underwent a lot of speech, physical, and occupational therapy.
Around the time Jacob turned 2 years old, we started fighting for genetic testing to be done. All basic genetic testing came back normal. All EEG’s and brain MRI’s came back normal, as well. Finally, we were able to do exome sequencing, an intensive genetic test that would take 6 months to find out the results.
On September 29th, 2014 our lives changed. Jacob had just recently had a modified barium swallow and a sleep study done so he could be put under for a spinal tap when we got a call from Neurology that his exome results were back and the result was abnormal. We waited in agony until his Neurologist called us that night with the actual results. “Glucose transport disorder” she said. One of the things she actually was going to look for in his future spinal tap! She explained the disease to us and how it was a new mutation on his genes and did not come from my husband or me. Jacob was to start the ketogenic diet as soon as possible.
One month later, we started the diet. It was really rough and still is in many ways, but Jacob has only had one minor episode since then and is full of energy. He is the happiest little boy and is always stealing people’s hearts. He finally learned how to walk this past summer and now we are getting an AAC device to help him learn how to talk. Jacob is a thriving 3.5 year old with a beautiful smile and determination like I’ve never seen. He goes to a special education preschool provided by our town and receives all his therapies there. He really loves school and has learned so much. We are truly blessed to have him in our lives.

Jaisa
When Jaisa was 18 months old, I was heading to my car leaving work when my husband called and said, “I think Jaisa just had a seizure.” I was in complete disbelief, and absolutely sure he was wrong considering he wasn’t even sure himself it was a seizure.
Later that same night, Jaisa indeed, had another seizure. It was initially very hard to recognize even with experience as a nurse. Jaisa has no prior history of any medical concerns except that she was delayed in hitting milestone, but the doctors were not concerned. Since she was born, she was a terrible sleeper and every night we would take turns being up with her all night. That night as I was in the living room with her she had another.
At 4am I called my friend and fellow nurse who I knew would be awake and described what I thought could possibly be seizures and she agreed. We took Jaisa to a local ER where she continued to have seizures every 2 hours like clockwork. Unfortunately, the ER brushed us off by saying they didn’t feel she was having seizures. I insisted she was and that is what we were seeing. At one point, Jaisa had a seizure while the ER nurse was in the room. I looked at her and said, “I’m telling you, this baby is having seizures.” She said, “I believe you.”
After some time she had a seizure that lasted long enough for the ER doctor to get there in time to see. We were immediately sent by ambulance to Hershey Children’s Hospital where an EEG confirmed seizure activity and she was given Keppra and admitted for the next 3 days.
Her seizures did stop while on Keppra with a rare breakthrough one. However, when she was 2, her neurologist at Hershey Children’s was concerned Epilepsy was not the best diagnosis given Jaisa’s delays in walking and talking and we were referred to their Genetics Specialist where testing gave us the diagnosis of Glut1 Deficiency. She was immediately referred to their dietitian and started on the keto diet. Five months after starting the diet she was walking and no longer on Keppra. She was also ordered physical and speech therapy. Once she was at preschool age, early interventions took over with physical therapy, speech therapy and occupational therapy.
Jaisa can now walk, run, go up and down steps, feed herself and has really done very well with diet and therapy combined. So much so that she was moved out of the specialized classroom and into a typical classroom. Her speech is still very much a struggle and she sometimes gets frustrated that she can’t communicate through speech. She is picking up sign language very well and will use that to assist her in communicating. The keto diet is challenging and comes with it’s own set of concerns for overall health but so far is the standard treatment for Glut1 deficiency.
It has been a long three years but she has come so far in that time. She is a sweet, happy girl who loves to play outside, rough house with her older brother, play with friends at school, and go to the beach. She continues to persevere every day no matter the hurdle put in front of her.

John
John was born March of 2016 with no indications of the journey we would embark on in the upcoming months. He was not meeting milestones aside from rolling over, which was a red flag but we remained patient as each kid develops differently. My mom instincts and educational background as a psychologist were telling me something might be off but could not pinpoint exactly what that was as no other symptoms were present. His first absence seizure occurred at 5 months old about a month after we started feeding him baby food. He had four seizures in the span of a couple of hours prompting us to head to the ER after the second one. The intake team saw the second set of them while assessing him and shortly after he was put on Keppra to manage the seizures. Over the next few months, he would experience break through seizures, both grand mal and absence, all while we continued to have tests run to rule out different disorders. He had his first MRI at 6 months old showing delayed myelination and his EEGs were abnormal. Thankfully at 18 months, his second MRI came back normal. We changed medications at the beginning of December 2016, which helped until we received the Glut1 diagnosis in February 2017 and started the ketogenic diet in April.
We are thankful that Dr. Phillips at Beaumont Royal Oak did not waste any time setting up genetic testing for John in December of 2016. We received the results in February 2017 a few months shy of John turning one. Dad and I were tested shortly after we received John’s diagnosis, which came back negative for both of us. It was hard to wrap our heads around how this happened but acceptance was the only way forward. We were scheduled with the diet clinic to start learning about the ketogenic diet. Family joined us at the first appointment to better understand the disorder and how the diet works. John was scheduled for a four-day inpatient admission at the hospital the first week of April, a week after he turned one. We were able to do a regular cake smash for pictures before being admitted. Starting the diet was trial and error because the team wanted to see if we could get to a 3:1 ratio but he was not tolerating the higher ratio. We left the hospital with John being on a 2:1 ratio and a lot of learning to do. Looking back, it has been a blessing that he started the diet at a young age because that is all he can remember. I will never forget having to make a keto friendly cake for his first birthday celebration. The cake tasted absolutely awful, and I can say we have come a long way coming up with some very appetizing birthday treats. My mother has taken on the challenge of coming up with new recipes with her most recent being keto friendly whoopie pies.
Our journey with therapies started in the winter of 2017 after being referred to the local school district for Early On Services. He qualified for OT, PT, and Speech therapy with a service provider coming to the house twice a week. We continued Early On at his current school district in 2018 then graduated to drive in services until he started three-year-old preschool in the fall of 2019. He has received school-based services from then until the present day. We have also participated in outpatient therapies when not in school. He continues to grow and learn every day, but we still deal with fatigue, movement episodes, and difficulties with temperature regulation. We are learning how to manage energy levels as he is always on the go and wanting to keep up with his peers and friends throughout the day. We are always figuring out ways to help him increase self-awareness of his body so he can advocate for himself when needed.
John will be entering second grade this fall 2023 in mainstream education with a lot of support from his teacher, the staff, and therapy providers. His classmates have been supportive at school, helping him with schoolwork and socializing. Everyone knows him by name and greets him with huge smiles as he walks through the doors at school. We spend every year revamping his plan of care to fit his current needs, which usually consists of creating a food schedule for him and making any adjustments necessary. While I am excited to see him move on to a new grade each year, it never gets easier explaining what Glut1 is to a new teacher and what to expect from him. Building a relationship with a new teacher can feel like a chore and by the time it feels solid, the school year is over. The school forms on the foundation’s website have been a very useful tool for conveying information about Glut1 and John. The two-sided sheet is given at the beginning of the school year, which allows them to start learning about him and ask questions if needed. John is academically behind with deficits in reading, letter/number recognition, and basic concepts. He struggles with sustaining attention and retention of the material he has learned. He continues to make progress in class but is behind compared to his peers. We have annual IEP meetings as well as meetings in between when his accommodations at school need to be revisited. The school system has been one of the more challenging environments to manage but we are finding resources to provide John with the best school experience.
While we continue to deal with various challenges, it has been a blessing to watch John grow into the kind, sweet, and fun kid he is. We ponder what the future will hold for him, but we know he has the greatest support system. There has been a lot to process since the beginning with each day bringing new challenges as well as amazing victories.

Jyl
Jyl was born on June 6th, 1992 at 41 weeks weighing in at 9 lbs. 10oz. She was born in the 95th percentile and, based on her measurements, seemed like a healthy baby girl. However, as time went on, her development just wasn’t progressing like we all expected. She didn’t compare to our friends’ children who were further along in their development than Jyl. Something was off… Our primary doctor would attempt to make a diagnosis ranging from Cerebral Palsy to Down Syndrome, but none of them fit. Over the next few months we would continue to dig, trying to find an explanation we could cling to, but still no answers.
At 18 months old, Jyl had her first Grand Mal Seizure. After receiving treatment at the ER, we were referred to Fort Wayne Neurology in Fort Wayne, IN. We finally started on a path towards a more accurate diagnosis. Dr. Ottinger, Jyl’s neurologist, did a typical neuro work up and could not see a reasoning behind Jyl’s seizure. He recommended a certain medication, but with no clear diagnosis, Jyl was weaned off the meds. Now, Jyl was 1 year old and still not standing or walking. She could barely say one word. Jyl was very big and developing at a slow rate!
As the years went on, Dr. Ottinger followed up through several “episodes”. Jyl’s eyes would bounce, head would drop, and lower limbs would become numb losing function. During this time, we also got in with First Steps for evaluation. They recommended OT and PT to help her physical development. Continuing to try and find answers, Dr. Ottinger asked if he could take a video of Jyl showing her difficulties that he would bring with him to a neuro convention. Unfortunately, upon his return from his convention, he still had no answers. It seemed that no one had any idea what was going on with Jyl. After reading a medical article, Dr. Ottinger had one more diagnosis he wanted to run some testing to establish. Jyl was approaching 4 years old and was not potty trained, only spoke in one word sentences, and walked very unbalanced. At this point of desperation, Dr. Ottinger told us he needed a spinal tap, but it had to be done during one of her “episodes”. Catching an episode for a spinal tap was no easy feat. Sometimes they only lasted 5 minutes, other times hours. There was absolutely no way of knowing. Finally, during the Summer of 1996, we made it to the Fort Wayne ER where Dr. Ottinger met us to perform the spinal tap. They sent the sample to Columbia Presbyterian Children’s Hospital in New York City. After an agonizing two week wait, we had a diagnosis. Jyl has Glut1DS.
Immediately, the doctors wanted Jyl to start a Keto Diet with a very specific ratio. It was very important for Jyl to become ketonic in order for her “episodes” to be prevented. Because of this importance, Jyl was scheduled for a 5 day hospitalization with water only until her body went ketotic. Luckily, it only took 48 hours. Once on the Keto Diet, Jyl’s development seems to progress more normally. She even began saying words to form sentences, which made all of our hearts burst with happiness. Jyl was developing, but delayed due to the crucial years she missed at the beginning while we were searching for a diagnosis. After her diagnosis, Dr. Hinton out of New York City flew out to see Jyl for an in-home evaluation. Shortly after, we were all flown out to New York for further testing and research where we discovered Jyl was only the 15th diagnosed worldwide with Glut1DS.
The diagnosis taught our family to learn how to live a different way. We measured every single bite that would go into her mouth. We had Jyl attend preschool both morning and afternoon class for the repetition. When she entered the public school setting, we made sure she had OT, PT, and speech all through her IEP. Outside of school, she attended additional OT and PT along with aquatic therapy. She even attended Red Cedar Center for horseback riding therapy.
By her Sophomore year she was entering into the menses stage of her life. Every slight hormone change caused her to go into an episode. She hardly completed a full day of school that year. We quickly noticed that the low carbs were NOT helping at all. We started letting her eat what she wanted and when she wanted. She seemed to be able to tell when she needed food for energy. We stopped all Keto, stopped all carb watching and just let her live a normal high school life.
In 2011, Jyl graduated with her class and received a certificate of attendance. She at this point is 19 years old. Out of high school, she was able to land a job at Martin’s SuperMarket. She loved her job! She worked for about 2 years until we stumbled upon a College Program that would be ideal for her skill set: Huntington University’s ABLE program. She started attending in 2018 when she was 26. Currently, Jyl is 30 and a senior in the program. She will conclude her college experience this school year and graduate in 2023!
Accomplishments
• 2011 – Whitko HS Prom Queen (Senior year)
• 2011 – Graduated HS with a Certificate of Achievement
• DAZZLERS
• National Champ – Cheer LTD – 2013
• National Champ – Apex Florida – 2018
• Coastal Cheer and Dance – Runner up – 2017
• MCDA Cheer and Dance – Race for the Rings Champion – 2019
• Cheer Max Champions – 2020
• JamFest National Champs 2016, 2017, 2019, 2020
• 2018 – Indiana Miss Amazing Miss – (Miss division winner)
• Competed in Nationals in Chicago, Ill 4 day event
• 2021 – Special Olympics
• Corn Hole State Champion
• 2nd Place 100 Meter Run
• 1st Place 400 Meter Run
• 2022 – Indiana Miss Amazing Senior Miss – (Senior Miss division winner)
• Competed in Nationals in Nashville, TN

Kamden
Kamden started having strange episodes at about 18 months old. He would lay on the floor and he couldn’t move, almost like he couldn’t remember how. As he started getting a little older the episodes increased in frequency and intensified. Not only would he lose the ability to mobile, he also lost the ability to speak and all he could do was cry, unable to open his eyes very much. We’d explored many specialists, done many tests and nobody even thought of Glut1.
After Kamden turned 3, we were sent to Childrens Hospital of Michigan for genetic testing, where we did many tests to see what was happening. When they finally tested the ataxia panel they accidentally discovered this mutation on the SLC2A1 gene and recommended we see a neurologist. We have been working with a Mott Childrens Neurologist, and also we’re seeing Dr.Pascual yearly to manage Kamden.
He is mild, and he experiences some developmental delays and movement disorders, but his course of treatment helps these things. Since diagnosis we have learned everything we can about Glut1 and what effects it can have on a person. It’s a lot to take in, but it helps to be as knowledgeable as possible about it. It’s such a rare condition that I often find myself educating people around me about it whether they are medical staff, family, or friends.
Things just make more sense to us now about what he’s going through and how to avoid episodes or make him feel better. In the future I just hope they keep studying and gaining more knowledge, and hopefully more treatment options will be available because the ones available now do have their struggles in the younger group of people who do have Glut1 DS.

Kinzlee
At the tender age of 4, we were aware of our daughter, Kinzlee, expressing intense pain in her legs and could hardly walk. We didn’t know at the time she had a rare condition and after a few years with doctor visits and physical therapy we were at our wit’s end. My wife thought to reach out to a physician at a center that takes blood and does a varitey of tests regarding possible genetic conditions. By the time my daughter was 5, we had found out that she was positive for Glut1 Deficiency and another disability that was paired with it.
Today, she is 6 years old and has been on the keto diet and on an oral medication to help her body. There are significant improvements with her walking and speech. She comprehends better but not everything she should. She also has various keto friendly snacks throughout the day. She loves playing with her sisters, we just need to continue to remind them that Kinzlee sometimes can’t control her reflexes and movements and it sometimes results in her hitting or bumping others. I do also notice her worsening when she eats something that is not on her keto diet and sometimes she will also throw up these because her body cannot process them anymore now that she is in ketosis.

Linzi
October 99…a daughter, a sister
And then it all began…
Linzi had rapid eye movements within 6mths..dismissed as wind,weak eye muscles!
All milestones failed to be be met…dismissed as “she’s a girl,don’t compare to when her 2 brothers achieved!”
Non verbal still at 2yrs,unable to crawl,eat solid food.
Having up to 100 absences in a day,then having full blown seizures but normal EEGs until one showed a sharp spike and epilepsy was confirmed.
Tested for Retts syndrome, Angelmans syndrome, Ataxic Cerebral Palsy … nope
Then she saw one last professor at Hammersmith hospital in London, they heard her story and she was brought in for a lumber puncture and advised not to start the anti convulsant drugs.
Within 2 days of the results Linzi was in Great Ormand Street hospital diagnosed with Glut1 DS and on the ketogenic diet…She took to it like a duck to water..Only three and a half years too late but.
Linzi unfortunately has her additional needs..due to her brain being starved of food it ceased to grow at a year and a half so she has acquired microcephaly, she is severely long sighted, cognitively 6yrs old,unable to read ,write and with no concept of time or day.
She use a kaye walker and self propelled wheelchair to get around.
BUT…
Linzi is a beautiful young 23yr lady that goes to college, enjoys drama,yoga,swimming, gardening, horse riding. She enjoys a good old chat and has a wicked sense of humour!!
Weirdly if she didn’t have Glut1 she wouldn’t be our Linzi…she’s loved by everyone ❤
THANK YOU KETOGENIC DIET…bring on many more years of weighed out liquigen emulsion, double cream and sugar free soya milk

Lochlan
When Lochlan was born she was a happy, healthy, full-term baby. She started to feel floppy around 6 weeks of age. Also at 6 weeks of age, she started having rapid eye movement where they shook really fast back and forth. She then began to have aberrant gaze saccades a few weeks later. By 4 months of age, she was having 100’s of seizures a day. She was diagnosed with Myoclonic Epilepsy by 5 months old and put on medication. When she was 8 months of age, she was on 2 different seizure medications, and ended up being hospitalized for an upper respiratory infection. Doctors began to run further tests to get to the bottom of what was going on.
At that point she was still having seizures all day and the eye movements she was having didn’t register on her EEG. They decided to do a lumbar puncture and CT scan to rule out Neuroblastoma.
It was discovered from her lumbar puncture that her glucose was low. She was immediately started on the ketogenic diet and, within 2 weeks, her seizures and eye movements stopped. She began to reach milestones. Later, her diagnosis was confirmed through genetic testing. Keto truly has changed Lochlan’s life! She is a happy, healthy, and energetic 8 year old.

Logyn
Hello Glut1 family! We are the Maggard family. Our daughter, Logyn, was diagnosed at almost two years old. I am Alexis and my husband is Joe. We are going to share our story with you all. Just three years before Logyn was born we suffered the loss of her big sister, Presley. Logyn was truly the rainbow after the storm and she was the glue that helped to heal our broken hearts. We were so excited for a sense of normalcy and a source of joy in our lives. In the beginning, there was nothing unusual. Both her and I were perfectly healthy during pregnancy and birth. It wasn’t until we got home from the hospital that we grew concerned.
Logyn was a very difficult, inconsolable baby more often than not. She had feeding issues both from the breast and a bottle, slow growth, and her developmental delays were evident very early on. We also noticed odd ticks, eye movement episodes, and blank starting spells that were brushed off as being something that she’d grow out of. By the time she was 6 months our concerns had grown. She was just meeting 3 month milestones and was behind in every developmental area. She spent most of her time in a dazed state.
We again voiced our concerns to her pediatrician who agreed that it was time for intervention. We had her evaluated and immediately began occupational therapy. Around 8 months old she had her first seizure. We made an appointment to see her pediatrician who ordered a 30 minute EEG, and from there we were referred to our first neurologist at Texas Children’s Hospital. To our surprise, the 30 minute EEG was completely normal. Thankfully we had videos of the first event to show the neurologist at our first appointment and she ordered another EEG, this time 72 hours as well as an MRI. We did the 72 hour EEG in February 2020, one month before her first birthday. The EEG came back abnormal, but her MRI was normal. Because the EEG results showed seizure activity we began Keppra to gain some control. It worked for a while but over time Logyn’s seizures started to evolve. She began having atonic seizures and was hurting herself constantly. Her medicine was switched from Keppra to Clobazam and another EEG was ordered, this time 96 hours, which again showed seizure activity. At this point, genetic testing was ordered. We completed the testing in November 2021 and in February 2022 we had a diagnosis of Glucose Transporter Type 1 Deficiency. From there it was a whirlwind. We were transferred to a new neurologist within Texas Childrens, Dr. Katyayan, and scheduled our first appointment with him as well as the dietician team in March.
March 2022 was a big month for our family!!! We started Logyn on the keto diet, and we also welcomed Logyn’s baby brother, Everett, into the world. Two weeks later Logyn celebrated her second birthday. By May, just two months after starting the diet, Logyn started walking, speaking, and her seizures were mostly controlled by the diet and Clobazam. Now, exactly one year later, she is thriving. She is a happy, intelligent, silly, and kind little girl.
They say that kids are resilient but Logyn Elise is that times one million. Everyday brings new challenges and we are navigating them the best we can. We still worry about Logyn’s clumsiness and residual seizures. She falls a lot and still wears AFO’s to help her walk. We are currently working with the dietitians to make modifications to her diet in the near future and hopefully gain more control. Now that baby brother Everett is almost 1 and is exploring solid foods, we are having to learn how to handle Logyn’s interest in his “normal” food since she is too young to understand. We know that this will likely be a lifelong challenge for us and for Logyn. Despite the challenges that this journey we are on brings, we remain hopeful. Our beautiful, bright girl has a chance at a mostly normal life. We are so thankful for that and the support surrounding us from family, friends, and medical team. We will continue to hope and advocate for more awareness to be made and research to be done for our Glut1 family. 💛

Lucia
Lucia was born on the fourth of July in 2023. It was only fitting that she would be our beautiful firecracker. When Lucia was 6 weeks old she had her first seizure. She began to sweat profusely, lost oxygen, and eventually went limp. This would happen three times again all resulting in a 911 call. In total we called 911 three times and spent a total of 15 days and nights in the hospital.
She was diagnosed with Glut1 by taking a saliva test via INVITAE. Three weeks later we get a phone call to come into the doctor’s office to discuss the results. Fast forward to another two weeks and we are at CHLA transitioning to the medical ketogenic diet. Lucia is now a 7-month-old happy baby who is testing age appropriately cognitively and is two months behind physically. We attribute her success to the early diagnosis and PT she has. The future is uncertain but so hopeful. We are so proud of our baby girl.

Macie
Macie was born in 1997 by C-section after a complicated delivery, but she had normal Apgar scores and all seemed fine for the first few months. At around her first birthday (about the same time she weaned from breastfeeding), we began to notice that Macie had episodes where her eyes would jerk upwards, and sometimes we would also feel or see slight jerking in her arms and legs at the same time. This always happened more frequently when she was tired or hungry, and they only lasted a couple of seconds. We mentioned these to her pediatrician, but he didn’t seem overly concerned, and the thought never occurred to us that these could be seizures. A few months later, Macie had a longer episode, more like a typical absence seizure. We took her back to the doctor, she had an EEg, and she was diagnosed with focal epilepsy at 22 months of age.
This seizure pattern continued, and Macie’s seizure count was in the hundreds daily. When she was 4, we decided that we wanted her to try the ketogenic diet, since the many medications we had tried were ineffective and the side effects unacceptable. We had to search out a neurologist just over 4 hours from our home to offer the diet to Macie, and he changed her diagnosis to idiopathic generalized epilepsy. She became seizure free as soon as she got into ketosis with an at-home induction, and medications were weaned within a couple of months. She took to the diet like a champ, and it was like a fog had been lifted and there was a new child underneath. We enjoyed this seizure-free period for almost 2 years, and then as we were beginning to talk about weaning the diet, the seizures returned. We tinkered with the diet for some time, and then ended up switching medical treatment centers again in hopes of being able to better fine tune the diet and regain control. Unfortunately, seizure freedom continued to elude us, and after another two years of adjustments and experiments, we weaned the diet completely when Macie was 8 years old.
The new neurology center diagnosed Macie with Doose Syndrome, or Myoclonic Astatic Epilepsy. Once we weaned the diet, we began the drug merry-go-round again, and Macie started having new seizure types. We found Diamox to be effective for the tonic and tonic-clonics, but nothing we tried seemed to help with the short absence/myoclonic seizures she continued to have. We were eventually told that the Vagus Nerve Stimulator was the only option left to try, so after getting second and third opinions we agreed to have the device implanted. It was turned off within a few weeks of her surgery because it seemed to aggravate her seizures. While undergoing video-EEg monitoring prior to the surgery, one of the attending neurologists diagnosed Macie with Lennox-Gastaut Syndrome. Macie’s younger sister, Maggie, has a history of complex febrile seizures, and we were also told that both of them could have GEFS+ (a familial epilepsy linked to the SCN1A gene – Maggie eventually had more genetic testing when her seizures returned at age 15 and was diagnosed with GRIN2A, a different rare genetic epilepsy).
We were never fully convinced that Macie had the proper diagnosis with Doose or Lennox-Gastaut, so we continued to search. I happened upon some information about Glut1 Deficiency, and the more I read about it, the more I thought she could have it. Her doctors were not willing to consider Glut1 Deficiency until her epilepsy center did some reshuffling and she was placed with a brand new neurologist. The testing process began with a spinal tap, then a fasting video-EEg, next a PET scan, and then DNA analysis for our whole family. All of her testing results were in a gray area for the diagnostic information for Glut1 Deficiency available at the time, so everyone was reluctant to make a diagnosis. Finally, in 2008 we visited Dr. Juan Pascual in Dallas and he immediately confirmed Glut1 Deficiency when she was 10 ½ years old. We started modified Atkins right away with a great deal of improvement for Macie, then later switched to a more classic version of keto with even better results.
Puberty proved to be a difficult time for Macie. She had more and worsening symptoms and we struggled to maintain ketones at levels high enough to be therapeutic. We tried fine tuning the diet with little success, so we eventually weaned keto and tried a carb-loading approach, including using corn starch. In 2014 we enrolled in the orignial C7 (triheptanoin) trial with Dr. Pascual at UT Southwestern. Despite our high hopes, it was not effective and we discontinued the C7 at the end of the 3 month study. We have revisited the diet a few times since, but we still have difficult time with achieving and maintaining ketones.
Currently, we manage her symptoms with frequent meals and snacks aimed at keeping her glucose levels stable. She continues to take Diamox and a few supplements that we think are helpful. Her seizures, stamina, and energy levels have improved, although we still see a few brief seizures in the morning before breakfast or when she is very tired or sick. Although we’ve never been able to recapture those two years of seizure freedom when she first started the diet, she is doing better now than any other time since. Her movement issues have always been on the milder side and more fine motor than gross, but we see a few more occasional muscle tremors or other signs of muscle fatigue as she has gotten older. She also complains occasionally of back and neck pain, which we can only attritube to some type of dystonia or spasticity flaring up. Macie received OT and speech therapy and intermittent physical therapy when she was younger, and has done physical therapy again to try to help with some of the back pain.
Macie is a happy, sweet, determined, and kind young lady, and she very much likes routine and predictability. She has moderate delays both cognitively and socially. She was home-schooled for many years but returned to public school in 8th grade. She has had a customized curriculum and the support of some great people at school and home, including her one-on-one aide in the classroom. Macie received her diploma in 2018 volunteers at our local elementary school. We look forward to opportunities she will have to meet other young people facing challenges and learning to adapt and flourish despite them. She especially enjoys and looks forward to our biennial Glut1 Deficiency conferences and being with dear friends she has made in the community.
The Glut1 Deficiency diagnosis has brought peace to our family with the explanation it gives for Macie’s symptoms, the difficulties in finding effective treatments, and the help it has given us towards acceptance of her special needs. It also gives us a place to focus our energy in trying to help her and a whole new family to share the journey. Macie has been such an inspiration and a blessing to our family and teaches many valuable lessons to all who know her.

Mackenzie
Mackenzie’s journey started when she was just 6 months old with her first grand mal seizure. Her second seizure came one month later. The seizures kept coming once a month despite medication. We saw her delays, but we were told she would grow out of the delays and the seizures by the age of two. Instead, at two she had her first seizure while she was sleeping and it lasted for over five minutes. This moment changed everything. Doctors began telling us she would likely always struggle with epilepsy.
After she had a seizure in front of her doctor during a test in April 2021, the team decided it was time for genetic testing to be completed for our family. In August 2021, testing showed that Mackenzie had mutated the SLC2A1 gene on her own. She had a PET scan in October 2021 to confirm. Mackenzie was placed on a Modified Atkins diet at this time. After some struggles with diet compliance, Mackenzie is back on her diet. She’s thriving and we are thrilled to see such huge developmental strides being made. Glut1 is tough, but Mackenzie is tougher.

Meredith
I was diagnosed with Glut1 Deficiency nearly 11 years ago, when I was 18 years old. I have been told that some of my earliest symptoms started in infancy, as I was lethargic and very low energy. Seen then as a blessing to my parents after having 5 older siblings and 1 younger, all high energy. No one thought much of my easy going behavior, until I was a little older and began having more symptoms. At the age of 2 I had my first movement disorder, which I began calling The Wiggles. I spoke in one word sentences for longer than anticipated, and hit major milestones on the low end of normal.
Once I started school I continued to be “easy going” and soon developed a learning disability. Once puberty began my symptoms became worse, not uncommon with Glut1, and I began to become much less social for fear of standing out as I became more aware of my differences. Once I started high school I couldn’t keep up with my peers, as all my friends were turning 16 and getting licenses I was left behind unable to drive for fear of The Wiggles coming while behind the wheel. About this same time our insurance changed and I got a new neurologist. In June of 2012, when I was 18, my neurologist attended a conference to learn about Glut1. She called us from this conference to schedule a Lumbar Puncture and in September of that same year, my Senior year of High School, I was officially diagnosed with Glut1 Deficiency.
Glut1 is a neurometabolic deficiency where glucose doesn’t reach the brain like it is supposed to, rather those with Glut1 have to use Ketones instead of Glucose. I am among only about 10% of Glut1 cases that do not have seizures caused by Glut1. There is no cure for Glut1 and the only known treatment is low carb diets. Many patients with Glut1 benefit from either a Keto or Modified Atkins Diet, but there are those whose symptoms may not improve, or even worsen, with the diet therapy.
I am now 28 years old, I attended college and received a Bachelor’s Degree in Sociology with a Concentration in Social Work and a Minor in English in 2017. I currently am a Special Education Instructional Assistant at a local elementary school. I am also recently engaged to my best friend, who helps me manage my Glut1 and cares for me when I am having a bad day. My symptoms have pretty much gotten better as I’ve gotten older, only showing up in high stress moments or if I am too tired. My fiancé and I are working towards me getting a drivers license someday so that I can drive myself to work and the grocery store when needed. Due to Glut1 being a genetic deficiency there is a 50% chance that any future children could be born with Glut1. Early diagnosis and treatment is key to success with treating Glut1.

Mia
Mia was diagnosed with Glut1 and began the medical ketogenic diet just before her first birthday. Her diagnosis was confirmed when genetic testing showed a deletion of the SLC2A1 gene. Mia turned three on March 5th of this year. In her three short years, she has experienced more than most – hospitalizations, blood draws, many many doctor’s appointments, weekly therapies and more. She has overall developmental delay. She struggles daily with things that would come naturally to most almost three-year-olds – learning how to walk, how to stand independently, learning to talk and communicate. She works hard each day to make small gains. We dream of a brighter future for Mia and the work being done by the Glut1 Deficiency Foundation gives our family hope that one day she will have more treatment options available and her progress will be exponential.
The McClendon Family (Mia, Rob, Jill and Miles)

Miguel
Miguel was diagnosed with Glut1 Deficiency at the age of 4 ½.
At 2 months of age, Miguel began having erratic eye movement episodes. Our family’s Pediatrician thought they could be seizures, but was unfamiliar with them. Little did we know, but this was just the beginning of a VERY long process to get the answers we needed for Miguel. After countless appointments with different Neurologists, Ophthalmologists, other specialists, EEGs, MRIs, weird eye violating tests, blood work, urine analysis and more, no doctor could give us any answers. The words, “it looks like nystagmus or come back in 6 months, maybe he will grow out of it” didn’t sit right with me. As time went on, other symptoms like episodic ataxia, excessive crying, myoclonus, and developmental delays presented themselves. As Miguel got older, he started missing milestones like crawling, walking, and talking.
Then in November of 2016, at the age of 2, Miguel had his first tonic-clonic seizure and then another 6 months later. In June of 2017, he was diagnosed with Epilepsy and put on anti-seizure medication.
Although we didn’t understand what triggered the seizures, Miguel would experience breakthrough seizures every 6 months which led us to another Neurologist who diagnosed Miguel with a rare neurological disorder called Opsoclonus-Myoclonus Syndrome (OMS). Our family was thrilled to have answers because it seemed like a match, but I started questioning it when the Neurologist didn’t want to do any treatment. Through my research I found there were limited experts on OMS and in the summer of 2018 we sought an OMS medical expert in Boston, MA. We had such high hopes to be put on a plan of action, to only be told it wasn’t OMS, but genetic. The Boston Neurologist sent us on a course of seeking out Mayo Clinic, which is where we went.
In a matter of weeks from our first appointment, Miguel underwent a week’s worth of various testing, which included a spinal tap. In March of 2019, we finally received a diagnosis of Glut1 Deficiency and started the Medical Ketogenic Diet shortly after. We started noticing changes for the better in Miguel 2 weeks after starting the keto diet and were so happy when the 6 month marker rolled around with no seizures.
Miguel is thriving on the diet and his brain is definitely trying to get caught up on all the building blocks he is behind on. I still need to pinch myself every time I share with people that Miguel will be 3 years seizure-free this November and is starting to talk and improve in all areas. Our family is so thankful for the Boston and Mayo Clinic Neurologists who actually listened and took action to set us on the right course of action. Glut1 Deficiency awareness in the medical field is extremely important. We are excited to share that our next appointment will be discussing the process of removing the anti-seizure medication, since it is clear that the medical keto diet is working.

Murray
At 4 months of age Murray was diagnosed with Glut1 Deficiency Syndrome. This diagnosis arrived 2 months after our first ER visit where Murray’s 100+ seizures/day were finally confirmed. We have been on a medical ketogenic diet for 3.5 years and seizure-free for almost 4 years. Every year we participate in the annual Glut1 Deficiency Foundation fundraiser because we were one of the lucky ones. The path to diagnosis can be long, painful, and full of endless challenges. Our medical team at CHCO were aware of Glut1-DS and the disease was on their list of possibilities early on. The effort and advocacy is so important to us as we remember what it was like to be among those searching for answers, just starting to put the pieces together. It’s a journey that never really ends. However, the journey can be made easier through awareness. More medical professionals need to be made aware of the telltale signs of the disease and how to treat it early in life. We also need more effective treatment options and those discoveries can be fueled by additional research in this area.

Negan
At the tender age of 9 months my grandson was diagnosed with Glut1 Deficiency. Before diagnosis, his mom and dad first noticed the rapid eye and head movements. Then one day while home with dad he had a seizure. He was taken to the local hospital and they ended up referring him to Children’s Hospital where he was diagnosed. He had to spend almost a week in the hospital where mom and dad learned how to prep keto meals. Since being on the keto diet he has not had any further seizures. We have noticed a big improvement with hand/eye coordination. He is talking a lot more and can now sit up by himself for short periods of time. He is in therapy and they are hopeful he will be crawling, standing up and couch surfing within 6 months. Our whole family is grateful to his team of doctors and to all the information we have received from the Glut1 community.

Olivia
My name is Olivia and I am 19 years old. I was diagnosed with Glut1 just before my 10th birthday. It took almost a decade, and moving from Connecticut to New Hampshire before a doctor finally properly diagnosed me. If not for my Neurologist, Dr. Richard P. Morse at Dartmouth Hitchcock Medical Center and Dr. Darryl C. De Vivo, I probably would not be doing as well in life as I am today.
Glut1 affects me mostly with seizures. I am on the Modified Ketogenic Diet and take anti-epileptic medications as well. I use a wheelchair for safe mobility and independence on days when I have seizures, hot days, and on marathon shopping days. As I have grown, and through OT and PT, my balance and ataxia have improved tremendously. I do pretty well in school and walked with my 2017 graduation class in June, I will continue in school until I am 21. I currently live in Enfield, New Hampshire with my family. I have a goal of living in an apartment by the time I am 25.
I enjoy many activities such as swimming, running, and horseback riding. I participate with Unified Skiing in the winter. Initially, I used a ski sled, but I now ski independently. I also play basketball on two different Unified Basketball teams. I would say that of all the sports I participate in, I excel in swimming and basketball.
I love all animals, however dogs and horses are my favorite. I have a Carin Terrier named Piper. One day I hope to have a horse. My family is working on getting me a seizure dog. I have some amazing friends, and my family and I are always doing something or going somewhere.
Update 2019
In June of 2017, I walked with my 2017 graduating class. I returned to school in the Fall at the Regional Resource Center (RRC) to complete my education there. It is a specialized school that integrates life skills with academics. I continued swimming, playing basketball, and skiing. I also have an internship at a local thrift shop.
In the Fall of 2017, the Modified Atkins Diet stopped working for me. I was having seizures all the time and was using my wheelchair all the time. The seizures became so bad that I was hospitalized. I spent one week in the hospital where we started the Ketogenic Diet again. It was amazing! My seizures were just about gone in only a week. I did so well that the wheelchair was a thing of the past. My Ataxia is gone and my balance is greatly improved. My independence is probably the best thing that has happened now that the seizures are under control.
In June of 2018 I graduated from the RRC and had the summer off to just have fun. Over the summer, I applied for a work study program called Project Search. Three hundred students applied, I was one of about 13 that were selected. It was a very proud moment for me and my parents.
In August 2018, I began my work study program at Project Search. By the time I graduate from the program in June 2019, I will have completed 3 to 4 different rotations, Ophthalmology, Pediatrics, Dietary, and Radiology. I will also have worked on my interviewing skills and general skills required to have gainful employment. I would not have been able to be successful or even be accepted into this program if my seizures were not under control. The Ketogenic Diet in its TRUE form has enabled me to achieve many goals I have set for myself.
In September of 2018, my G-Tube was removed. It had been in place for 10 years. I now have two belly buttons! I remain mostly seizure free. I have some small breakthrough seizures if I drink more liquids than I am supposed to or have more caffeine than usual (I LOVE coffee).
In February 2019, I turned 21! We had a party and I ordered a Dirty Grey Goose Martini with NO Vermouth. We got the green light from my Keto Dietitian I just had to take 20 mls of MCT afterward. I had no seizures. A success!
I am in my last rotation at Project Search and I look forward to graduating in June of 2019. I will also receive a REAL 100% High School Diploma. This is HUGE! It will help with my employability. I plan on taking a pet grooming class over the summer where I will receive a Certificate as a Pet Groomer.
I hope to get a part time job and also start taking riding lessons again over the summer. I’m also thinking of taking a belly dance class and catching some concerts. We have a summer music series at local parks here in New Hampshire that are amazing. We live in the Lakes Region of New Hampshire, so I will be out on my Kayak as much as possible this summer as well.
I am living my best life. It has been a struggle to get here, and sometimes it still is. I am at the point in my life where having a partner would be nice. But, I have amazing friends and family and I have a full life. It will happen when it happens.

Paisley
Paisley is currently 14 years old and was diagnosed with Glut 1 at age 8. She is your typical teenage girl who loves to chat with friends, plays games on her phone, and be silly! Paisley‘s medical journey, however, began in utero. I was diagnosed with HELLP syndrome at 31 weeks gestation which led to Paisley being born preterm at 32 weeks. All went well in the NICU, she thrived and grew enough for us to take her home at 1 month old. Feeling as though we had survived “the hard part” I was crushed when her pediatrician ordered her first EEG at 3 months old. I expressed concerns about what I thought was an overactive “startle” reaction. The EEG proved that they were myoclonic seizures.
As Paisley grew, her seizures became numerous. She was having hundreds of clustered myoclonic seizures a day. Her neurologist at the time was baffled. Numerous medications were tried, numerous EEG tests, MRIs, all giving us no answers. She was then given a diagnosis of “generalized epilepsy.”
As a mother to two previous children, I saw, too, that her development was not on par. She was slow to sit, stand, crawl, walk, and talk. Even when she started to walk, her daycare teachers often observed her lying on the ground “resting” instead of playing with others. They often had to carry her from one room to another because she would cry and refuse to walk. Her neurologist and pediatrician assumed this was stemming from her preterm birth and her seizures, but that she would eventually catch up in her development. We were then offered occupational, speech, and physical therapy. (These were continued until she reached school age).
At three years old we were able to try Valproic Acid (Depakote) and we finally saw relief. However, at this time she then began having what we now know to be migraines. She would collapse to the ground in pain rolling back and forth crying until she would finally vomit and then sleep. Her neurologist increased the Depakote in hopes that it would alleviate her migraines. They did become more infrequent, but they were never gone.
We found as parents that we began guarding her sleep. We figured out that her seizure clusters were always worse when she was tired. We never stayed out long as a family at events, we were always home in time for Paisley to get a good night’s rest. We were adapting our family’s routine to support her comfort because that felt like the only way to get her some relief. Still, though we had many questions and no answers.
Paisley began public school at age five and I requested special education testing, knowing that she was behind her peers. She did qualify and began resource classes in kindergarten. From kindergarten to third grade she struggled to learn, she struggled with energy, but she was a happy child and did all she could to keep up. She was still having myoclonic jerk and absence seizures very often throughout every day.
At 7 years old, new symptoms began. Paisley began having movement episodes. I first thought she was being silly because we lovingly refer to her episodes as her “drunken sailor” walks. She would walk as if she was on a moving ship in the sea trying to keep her balance. These movement episodes increased over the months and she would have to be carried when they were at their worst. They would happen at school, and teachers would have to carry her to the nurse. We did not know it at the time, but the episodes of ataxia, dystonia, and chorea were about to lead us to a new diagnosis. These were not seizures. This was new.
One thing we noticed about Paisley‘s diet through the years was that she was naturally eliminating carbs from her meals. She asked for and craved very high-fat foods. She would request bacon, sausage, and hot dogs, but never wanted bread or mac and cheese like others her age. I mentioned this to her neurologist who then referred us to a colleague who specialized in metabolic disorders thinking this (along with the new movement episodes) might lead us to answers. It only took one visit with this specialist for him to make the connection between her diet requests, her seizure pattern, and her movement disorders….he had a genetic test done suspecting GLUT 1.
The results came in and we confirmed at age 8 that Paisley had Glut 1 Transport deficiency. In a way, it was a huge relief because we finally had answers. However, we had just begun a new journey. We were referred to another neurologist specialist and began to do all we could to find her relief. Without question, the ketogenic diet needed to be her main form of treatment. This, however, proved to be disastrous for her. Within a year of this diet, we realized that we had caused food trauma. She was not able at that age to understand why we would change her lifestyle so much. She felt alienated at school because her friends were eating anything they wanted. She began hoarding and stealing food where she could. We placed locks on our pantry and cabinets, but she would discover ways to work around her food boundaries. We could not continue this way for her mental health’s sake.
Her neurologist then referred us to a c7 oil trial with Dr. Pascual. We spent the next 9 months trialing this supplement. Dr. Pascual and his research team are truly top-notch and the research done with them has been very helpful. This treatment, however, would not be one we would continue as it caused her much gastrointestinal distress and we did not see significant improvement.
That brings us to today. At 14, Paisley is on medications to control the seizures, adhd, and movement episodes. She has begun to regulate her energy use to make the most out of her days. She naps often, snacks often, and opts out of many social activities, especially if they involve a lot of walking and or heat. She still has occasional seizures (including tonic-clonic) and movement episodes. We feel fortunate as she certainly is not as severe as many others with this condition. We know that she is still on a journey of figuring out how to live with this disorder. We just do all we can to support her in making good decisions for her health. We hope that as she ages she will come to terms with her condition and find ways to make the most of life.

Reed
Reed was born at 34 weeks due to IUGR and Preeclampsia. He presented initial Glut1 symptoms that were mistaken for common issues related to preemies with IUGR. He was delayed in most milestones but we were told he’d catch up by the age of 2 and go on to live a “normal” life.
Reed‘s journey with Glut1 began on 7/25/2021 shortly before turning 2 with a tonic-clonic seizure. He stopped breathing for about 2 minutes (which felt like 20), turned blue and stiff. Not something any parent should ever have to experience. It was a moment that changed our family forever.
Diagnosis: 8/12/2021 – Glut1. I was shocked, angry, sad, and also grateful for an answer. Until the diagnosis we were almost certain the genetic testing would be normal. My husband and I did IVF and our embryos were genetically tested! I’m still very much struggling with coming to terms with it all. How did this happen?
Progress: 9/7/21 – Reed started a strict medical keto diet and received a g-tube placement due to continued swallowing issues. We quickly saw significant improvement in Reed‘s overall health and wellbeing. To date, he’s gained 8 lbs in 4 months, is walking and improving in all areas of development. A medial keto diet is the only treatment for Glut1 currently. It gives Reed‘s brain energy through ketosis and helps in reducing the occurrence of seizures.
Updated Diagnosis: 1/7/22 – After visiting Dr. Juan Pascual, a leading Glut1 doctor and expert of the disease, he took a deeper dive into Reed‘s genetic deletion and identified 2 other notable areas which could cause additional symptoms and issues.
Today, Reed‘s updated diagnosis is “Contiguous Genetic Deletion Syndrome” and Glut1 is a part of it. We continue on the path of learning how to best support Reed through testing, ongoing therapies, a keto diet, and research. In addition to a lot of love and prayers.

Rian
When Rian was 6 months old, we believed she had a seizure. Doctors at the time said that it was reflux. We were in disbelief and that was not a good enough answer. Her Pediatrician had ordered an EEG and the results came back normal. Then 2 weeks later, she went cross-eyed for about 3 minutes. That raised a huge red flag for us. We then went to see a Neurologist who ordered a MRI which also came back normal. Rian would go through these periods of time to where she could hardly sit up. Rian was a happy baby, but at the same time, not reaching milestones like her peers. We were concerned parents and we wanted answers.
Rian didn’t start pulling herself up till she was about 14 months old and did not fully walk until after her 2nd birthday. She had tubes put in her ears around this same time and we were hoping this would help, but no improvements were made. When she did walk, it was with a wide unsteady gait, as if she was drunk. By this time, Rian’s left eye started going lazy so we took her to an Ophthalmologist who prescribed glasses. We were hoping this would possibly help with her balance. During this period of time, we started Earlysteps to try and help Rian with her milestones and reach goals. We then saw a Developmental Delay Specialist who could not put his finger on what was going on with our daughter and a Geneticist who ran every possible test. All tests that the doctors were running were all coming back normal and this did not sit well with us.
We decided to take her to a Neuroethology who ran several vestibular tests on Rian and ordered a CT scan of her inner ear. When these tests came back, they were normal as well. We still didn’t understand what was going on with our baby. There were days where she would be okay and others it would make you want to cry, scratch your head, and fight to find out what was going on.
We then saw a 4th Neurologist for another opinion, hoping that there would be something the other doctors may have missed. At the time he could not put his finger on what was going on. Rian started having involuntary movements in her body along with her unsteady gait and she would go through a period of time during the day to where she could hardly stand up and function. Finally, we were at our wits end and decided the only thing we could do at this point was to get the whole exome sequencing test done.
We are beyond happy to say we finally received a diagnosis for Rian. It had been a long journey. Without this test, we may have never been able to get a diagnosis for our sweet girl. She has been a trooper through all her testing and doctor appointments. We were able to send the whole exome sequencing genetic test in November 2015. At the time we believed it would take three to five months to get the results back. On Friday, February 5, 2016 we received a call that has now changed our lives. Rian has finally been diagnosed with GLUT1. This is the answer we have been waiting for since she was 6 months old.
Since her Diagnosis
Rian is on a modified Atkins diet, which limits her carbohydrate intake to 20 grams per day. Her Neurologist does not recommend using the ketogenic diet unless her symptoms worsen. So far, the modified Atkins diet has helped her improve in many aspects. Her speech has drastically improved and she is now able to walk without falling over multiple times. Within two weeks after starting the diet, she began to pedal her tricycle and just recently she began jumping. She still receives physical therapy, occupational therapy, and speech therapy. She is the most fun-loving and happy little girl. We are so blessed to have her.

Sam
Sam was born in March 2001. His mother’s pregnancy was normal and Sam was induced and delivered normal at 41 weeks. His first 6 months were uneventful, although mom noted he developed more slowly than his brother (2years older).
Sam began Physical Therapy with First Steps at 9 months. OT and Speech were added over the next few months and continued through middle school. At about 9 months, Sam had his first eye-dancing episode. A few months later, he had a drop seizure (lost all muscle coordination). Both episodes sent Sam to the emergency room for various tests from MRI to CAT Scan, blood and urine samples. All tests came back normal.
Sam changed neurologists and Dr. Garg at Riley Hospital (since deceased) in Indianapolis recommended a spinal tap and consulted with Dr. De Vivo at Columbia.This resulted in our first trip to New York City when Sam was age 2 to confirm his G1D diagnosis. We began the Ketogenic Diet and Sam is still on it to this day. Although he only had two known seizures prior to diagnosis and the ketogenic diet, Sam’s disposition changed dramatically after being in ketosis. He was much more content.
Sam began school in Early Childhood with an IEP at age 3 and was promoted into Kindergarten with his peers. Sam did not speak until age 4 or walk independently until age 5, but his comprehension was solid. Sam continued to have PT, OT, and Speech in school and hippotherapy year round outside of school since age 5 years. With his IEP, Sam had a full-time aide through elementary school. When we moved to Texas at age 10, various school therapies were dropped, but he continues with Therapeutic Riding and Brain Therapy, as well as tutoring in the summer. Sam also receives regular chiropractic care.
Sam continued in mainstream schools, although when we moved to Texas we decided to have him repeat 3rd grade. In High School, Sam was in regular classrooms with some inclusion support and accommodations. The only class he has in Resource is English/Language Arts. He enjoys and does well with his academics, but he does spend extra time studying.
Sam sings in the high school and church choirs. He also started his first paying job and works 3 hours once a week in the summer and once a month during the school year. He enjoys lifting weights and works on coordination training by punching and kicking a heavy bag. Sam works out 3 to 4 times per week along with his horse back riding. Sam also feeds and walks the family dog as his regular responsibility. He is becoming more and more independent each year. Sam also does brain training on a weekly basis to assist in the development of his fine and gross motor skills, balance, and agility. Sam is still on the ketogenic diet and is growing taller each year. He still has somewhat limited balance and coordination. Sam had no remarkable G1D seizures or events until about age 16, when he had significant growth spurts and began maturing through puberty. He has had occasional PED (physically induced dyskinesia) events, but these are short and he recovers quickly with 10 to 15 minutes of rest and a high-fat snack. am is a delightful young man and we are confident that he will have a bright and productive future.

Tessa
Tessa began having seizures at five months old and she has just recently been given the diagnosis of Glut1 at age nine. For the first five years of Tessa’s life, she was on twelve different seizures medications that never made a difference with her 30-100 seizures per day, and resulted in terrible side effects.
She had unexplained aggression, was not able to talk, and her attention could not be held for more than a few minutes. At age five, we decided to try the ketogenic diet which completely stopped her seizures from the first day of the diet. While on the diet, Tessa became a completely different little girl. She began talking and interacting with us, her aggression disappeared, and it seemed like a fog lifted off of her. After two years of being on this very restrictive diet, we tried to wean her off of it, but her seizures returned.
After two more years of struggling with the diet and seizures, we made an appointment with a metabolic specialist at Cleveland Clinic Hospital. With the symptoms we were describing to the specialist, he immediately felt she had Glut1 Deficiency and ordered several tests. One of the tests came back seven weeks later as positive for the Glut1 Deficiency gene mutation. While relieved to finally have a reason for Tessa’s seizures, we initially felt very angry that this diagnosis was not made at a younger age. For five years of her life her brain did not get energy to develop; but we realize that we can’t change that now. What we can do is raise awareness so that children with Glut1 are diagnosed earlier and help fund research for a cure.
Tessa is the most amazing and giving little girl, and we thank God everyday for bringing her into our lives. She never complains about being on this very restrictive diet, she never complains about having up to four therapy sessions a day, and yet she will worry and become upset if another child or adult is hurt, sad, or upset. She has helped us to understand the true meaning of compassion and love.
2011 update
Tessa is now 10 years old and continues on the ketogenic diet at a 3:1 ratio. She still has some brief absence seizures every day, but she seems to simply work through them and carry on. We have discussed adding on a seizure medication, but we are VERY apprehensive due to the side effects she experienced from the 12 meds she tried prior to starting the ketogenic diet.
Tessa continues to learn, but at a slower pace. She will again be in a 6:1:1 class this fall with the same wonderful teacher she had last year. Tessa made great progress with her reading this year, though math continues to be a challenge for her. Her teacher and all of her therapists say she always puts forth good effort and we couldn’t be more proud! Tessa also made great progress with learning to swim this year, though jumping in the pool is her favorite part! We continue to be amazed by Tessa’s compassion, empathy, and sense of humor.
We had another fundraiser for Glut1 in June, which was a huge success. We are hopeful that Glut1 gets more funding for research, and want to be part of raising awareness.
2018 update
Tessa is now 17 years old and she continues to brighten our world with her sense of humor and kind spirit. Tessa transitioned to the Modified Atkins Diet (MAD) in 2017, and is enjoying the less restrictive meals and increased variety. We travel to Baltimore, MD to the John Hopkins /Adult Dietary Therapy Clinic twice a year now, where they advise and support us . After doing all things keto for 11+ years, the transition to MAD was a difficult concept – mostly for us, her parents! We still haven’t given up measuring and searching for new recipes, but the more relaxed approach to meal planning has been a blessing.
We continue to go to Dallas, TX once a year to see Dr. Juan Pascual. Tessa took part in Dr. Pascual’s Triheptanoin (C7) Study in August 2017, but we now know that the oil is not a helpful treatment for her. We still have a lot of hope that there will be other options for treatments and eventually a cure.
At around age 14 Tessa began having movement episodes, that still continue. Being a teen and dealing with anything that seems at ALL off the “norm” is tough; and so this part of Glut1 is pretty hard on her. There are times when she needs to use a wheelchair as her feet have little control or times when she can’t control hand or head movements. Emotionally and physically this is her biggest challenge. She talks with a counselor at school a few times a week to try and work through these feelings as well as learning self-advocacy.
Tessa is in a wonderful life skills classroom in school and will be in that program until she turns 21. She has developed a great social network of friends who do extracurricular activities together throughout the year. Tessa enjoys bowling, girl scouts, swimming, and though she doesn’t enjoy shopping she definitely enjoys new clothes! Our family has grown since 2011, and Tessa now has a niece and nephew that love her as much as she loves them! Tessa’s sisters live only a short distance away and she enjoys spending as much time with them as possible.
As everyone knows that is on this journey, there is so much more to our story than what can be written here, but that’s a brief update on our beautiful girl. We feel so fortunate to have the G1D community to ask questions, share our struggles as well as our triumphs. We’re very thankful to the Glut1 Deficiency Foundation, the volunteers that accomplish SO much, and the donations that make such a difference. We cheer whenever we read stories about families getting an early diagnosis. We get excited as we read every tidbit of progress towards research. And we cannot wait to have Tessa be a part of the next conference to meet up with others going through similar things. ALL of these things are occurring because of people that care and the Glut1 Deficiency Foundation. XOX

Victor
Victor was diagnosed in 2019 he was 5 1/2 years old.
We are French living in Boca Raton FL.
Victor had his 1st seizure on my birthday when he was 18 months old and his 2nd 5 months after.
He was on Keppra for 2 years ( before we knew what is was) but the sides effects were too much for him ( losing balance, feeling like he is drunk, weakness).
As soon as he was diagnosed, this group helped me so much by finding the right medical team, answering questions in minutes.
We started the MAD at 20g of carbs a day and we did it for at least 3 years.
We were lucky enough to do Dr Pascual clinical trial in Dallas, but unfortunately it was not a good fit for Victor.
Victor is now 8 years old, he is Mild, he is not on any diet.
He was so frustrated with the diet and noticing a lot of changes that I stopped.
He is not crazy with sugar, he prefers salmon sashimi and sushi than a Mc Donald
Victor is in 2nd grade in public school, he spends 1 1/2 hour per day in Special need class for reading and math and the rest of the time he is on mainstream with a paraprofessional.
I found an aftercare program YMCA that works for him ( special need program) and he loves it.
He is practicing karate and swimming lessons.
His symptoms are
ADHD ++
Trouble sleeping
He had 2 seizures, and one hemiplegic migraine (super scary at 1 st but I remember a post about it.
I sent the symptoms and in minutes someone answered and help me going true what I had to do)
Victor is a joyful and loving boy.

Vivianna
Vivanna will be 5 this year (2023), she is the toughest little girl I know and special in so many ways!
She was born full term in 2018, and spent about 1 week in the NICU for low blood sugar. The hospital was able to get her sugar stabilized and sent us on our merry way. Around January 2019 I started to notice these strange “pumping” motions in her arms and legs (especially right before she was nursing). These movements didn’t seem right to me, and I started to feel like something was wrong. On Valentine’s Day of 2019 (6 months old), we took Vivianna to the hospital to check on these movements. The doctors were able to confirm that she was experiencing seizures.
My heart was broken to see my first baby being poked, prodded, and tested. We were given a diagnosis of Epilepsy and discharged with no reason why and no outlook into what the future would hold for my daughter. I spent countless days and nights worrying about her health, and always felt like there was something more than Epilepsy going on. Well, my “Mom Intuition” was right. In January of 2020 (17 months old) after completing a genetic test, Vivianna was diagnosed with GLUT1 Deficiency Syndrome.
Vivianna was delayed in hitting her milestones. She was a “quiet” baby, she didn’t start babbling until she was about 10-11 months old. She started crawling at 1 year old and started walking at 2 years old.
On Valentine’s Day of 2020 (18 months old), Vivianna was discharged from the hospital after spending 1 week initiating a strict Medical Keto Diet. That week in the hospital was the most heart wrenching experience in my life. To this day, it still remains an experience too difficult to speak about. However, initiating the medical keto diet has been the best thing for Vivianna, we have really seen her flourish!
Vivianna is in her 2nd year of attending half day preschool 5 days per week. She receives speech, physical and occupational therapy. She is able to talk, sing, climb, jump, run and dance (she LOVES to dance!). We are so grateful for the support of the Glut1 Foundation and hope to see many breakthroughs in the years to come!
We want to hear from you! Share YOUR story with our community! You never know who needs to hear it. We feature stories on our social media pages along with our website. Fill out our online form or send inquiries to [email protected].